Gene function prediction#
To find the function of an unknown protein, we would thus look for such conserved domains and compare amino acid sequences. There are many databases we could search to find similar sequences, but rather than just using the largest possible, we should consider the quality of evidence provided by each. For instance, RefSeq is a more carefully curated set of sequences than GenBank as a whole. Further, evidence of an actual protein sequence and function is better than evidence of only a transcript. While a transcript means that the gene is transcribed into RNA, this RNA might just have regulatory functions and not being translated into a protein necessarily. Many resources exist that aim to collect the best quality information about genes and proteins and we are going to cover a few below.
UniProt#
A useful resource for protein sequence and function information is the database UniProt, a collaboration between the European Bioinformatics Institute (EMBL-EBI), the Swiss Institute of Bioinformatics (SIB) and the Protein Information Resource (PIR). Within this project exists Swiss-Prot, which is manually annotated and reviewed, and UniRef, with protein clusters based on sequence similarity thresholds.
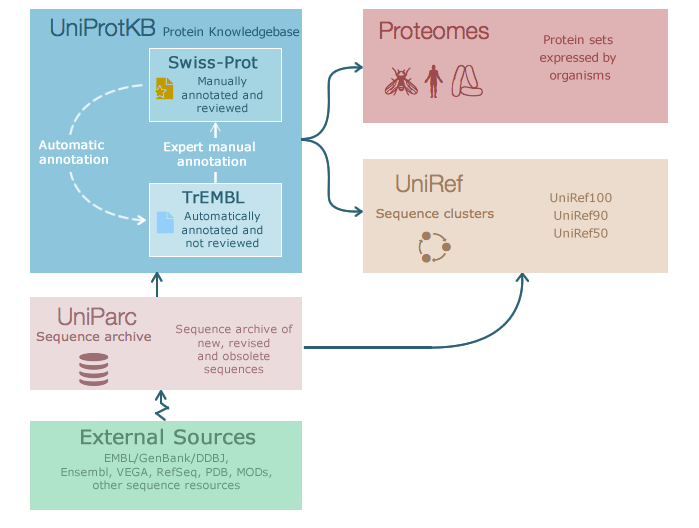
The website allows you to search by text, such as gene name or organism, or by sequence with BLAST. As a brief introduction, let’s look at the entry for a well known bacterial protein, DNA Gyrase subunit A or gyrA:
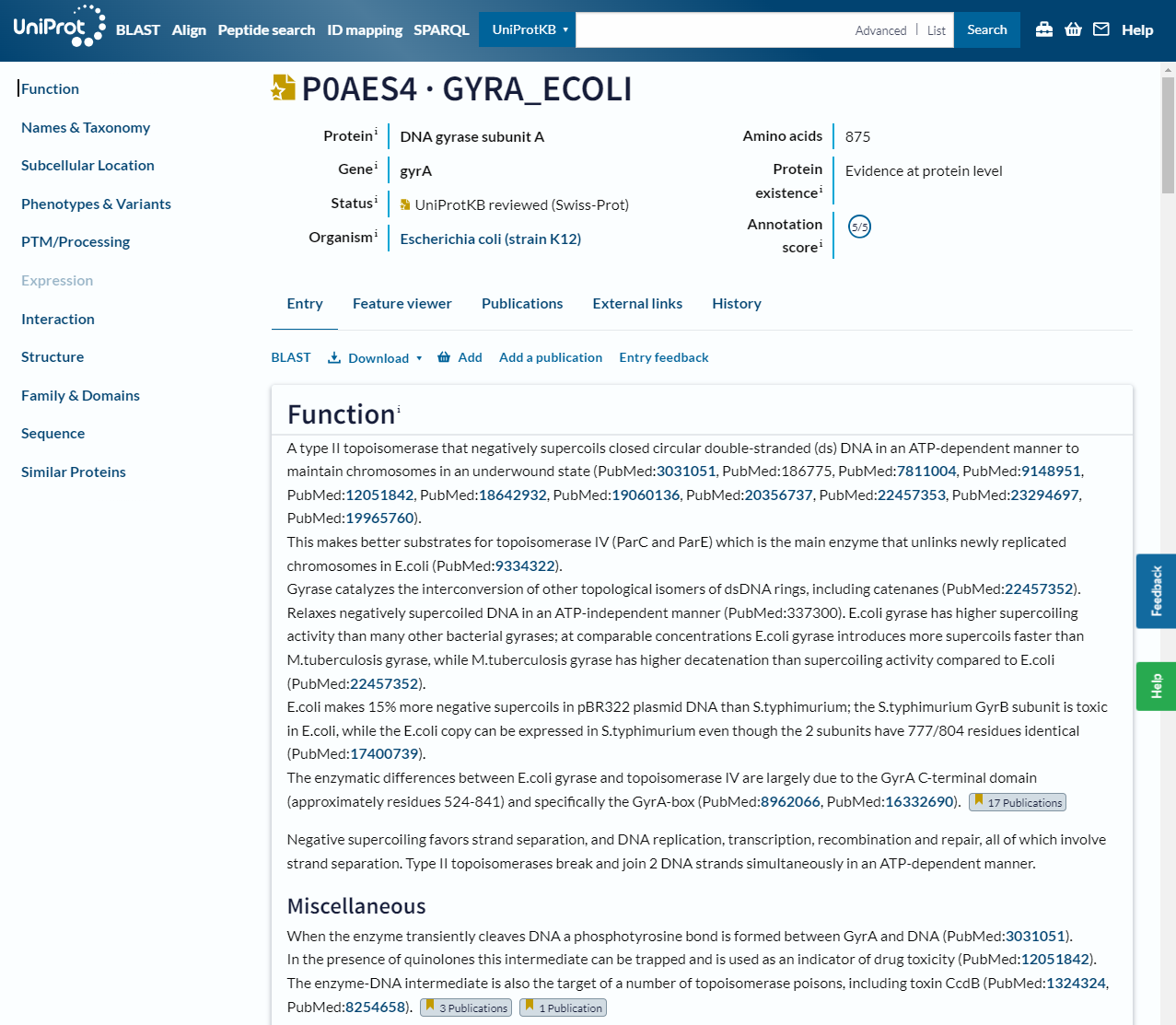
In this header you can see the gene name and organism it is from, as well as the annotation status, which gives you an idea of how much evidence there is for this particular annotation. There is also a summary of the functions of the protein with links to the scientific papers that support these statements. On the left is a table of contents for the rest of the page:
Names & Taxonomy: lists standard names, any alternative or historical names, the organism and its taxonomy
Subcellular location: shows you where in the cell the protein has been located, if evidence is available
Phenotypes & Variants: describes phenotypes and disease status associated with the protein and possible mutations of it, as well as drugs and chemicals it interacts with
PTM/Processing: lists post-translational modifications and processing of the molecule
Expression: information on the mRNA and protein levels in the cell or tissue
Interaction: describes the quaternary structure of the protein and interaction with other proteins and complexes
Structure: information on the tertiary and secondary structure of the protein that may include 3D structures from experiment or prediction
Family & Domains: information on sequence similarities with other proteins and its domains (we will discuss this more in the next section)
Sequence: the amino acid sequence of the protein as well as known variants and database listings
Similar proteins: links to other proteins in the database based on percentage identity
Cross-references: information from other databases (a summary of links in other sections)
Entry information: metadata about the protein entry itself
Miscellaneous: information that doesn’t fit into any of the other sections
As you can see, a vast amount of information is available about a single protein and UniProt does a good job of collating it all for easy access.
When it comes to annotation, we can use UniProt as a database for alignment of our unknown genes. We could also use SwissProt as a narrower but more trustworthy database, since it is manually curated. If we wanted to use broader but more hypothetical information, we could consider looking at protein domains.
InterPro#
In the “Family & Domains” section for Gyrase A, one of the databases linked to is InterPro, also run by the EMBL-EBI and indeed based on the data in UniProt. InterPro is database used for classification of protein families, thus it groups proteins containing similar domains and important sites. InterPro is an overarching database which uses models to predict protein function based on different databases (so-called member databases). If you click on Browse and then on By Member Database you will receive an overview of existing databases used by InterPro (including the commonly used Pfam and Tigrfam databases).
InterPro is a resource that provides functional analysis of protein sequences by classifying them into families and predicting the presence of domains and important sites. To classify proteins in this way, InterPro uses predictive models, known as signatures, provided by several collaborating databases (referred to as member databases) that collectively make up the InterPro consortium. A key value of InterPro is that it combines protein signatures from these member databases into a single searchable resource, capitalising on their individual strengths to produce a powerful integrated database and diagnostic tool. InterPro further provides its entries detailed functional annotation as well as adding relevant GO terms that enable automatic annotation of millions of GO terms across the protein sequence databases.
InterPro integrates signatures from the following 13 member databases: CATH, CDD, HAMAP, MobiDB Lite, Panther, Pfam, PIRSF, PRINTS, Prosite, SFLD, SMART, SUPERFAMILY AND TIGRfams (the InterPro Consortium section gives further information about the individual databases).
The member databases use a variety of different methods to classify proteins. Each of the databases has a particular focus (e.g. protein domains defined from structure, or full length protein families with shared function).
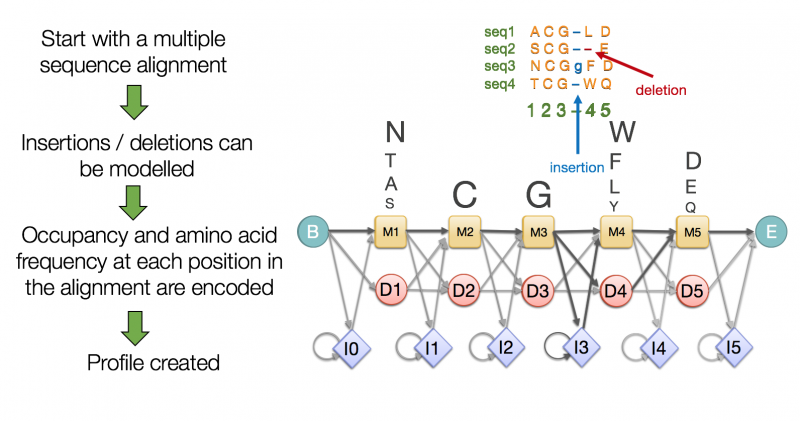
For example Pfam uses the principle of protein domains to define members of a protein family. A protein family is a group of proteins that are evolutionarily related to each other and therefore highly similar. A Pfam family consists of a small set of representative members, a multiple alignment of their sequences and a profile Hidden Markov Model (HMM) built from the multiple sequence alignment (MSA).
A Pfam HMM is a statistical model that encodes the likelihood of each amino acid at each position along with the likelihood of an insertion or deletion. You can take any sequence and score it with such an HMM to determine whether it is likely to be represented by the model or not, and therefore whether it is likely to be a member of that particular Pfam family
Let’s look at an example entry in InterPro the DNA gyrase C-terminal domain, beta-propeller which is a Pfam entry.
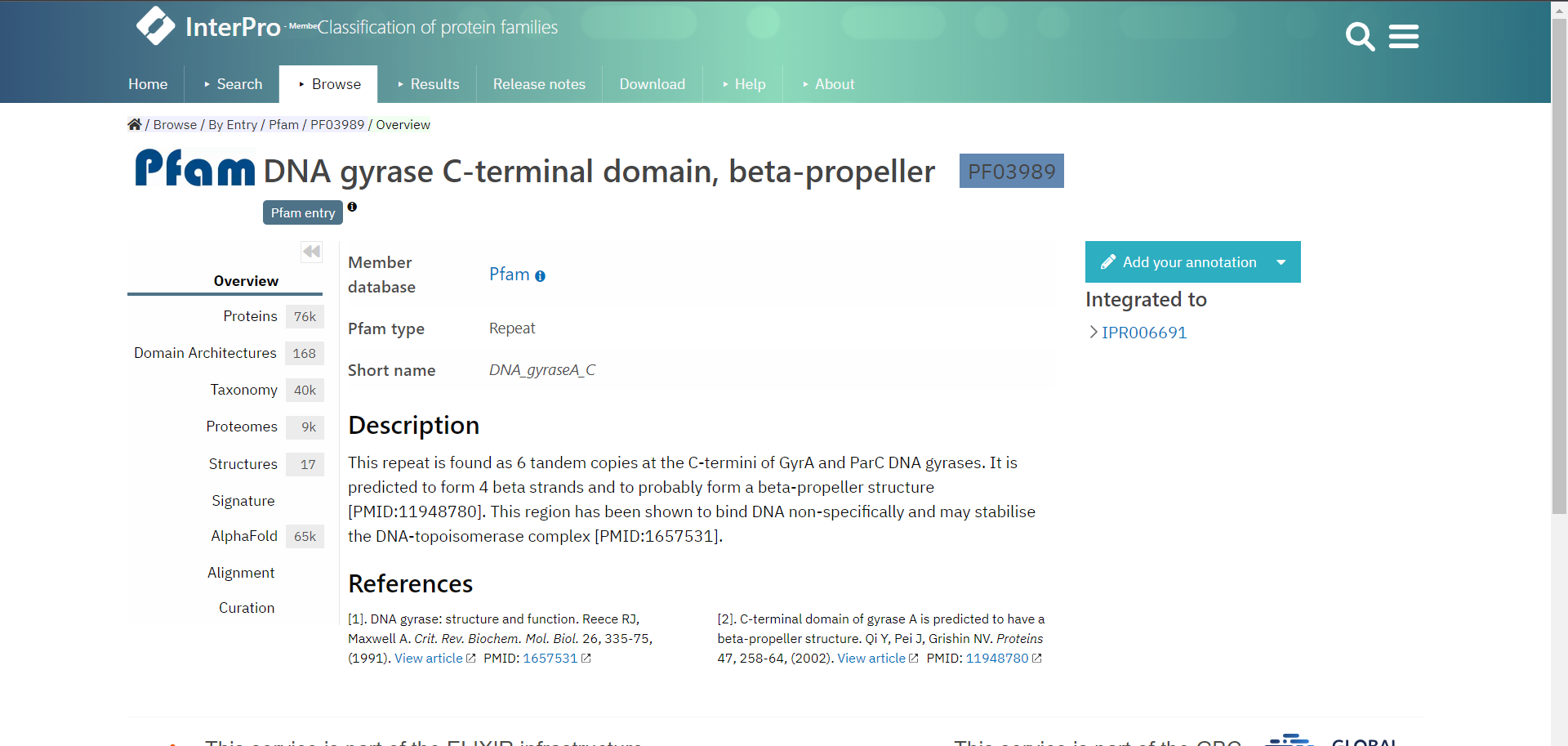
InterPro provides several useful pages of information accessible from the menu on the left, depending from which database your entry is from, the pages can vary. It is therefore the best if you explore the InterPro website yourself. However, each entry will have an Overview page which tells you from which database the entry is from and describes the entry and gives references literature to support this.
To use Pfam effectively, there is a suite of software called HMMER that can be used to both produce and search HMM profiles.
HMMER#
HMMER is available here, and we have made it available on the module system (ml HMMER). We will quickly show you how to build and search with an HMM.
To construct an HMM from a multiple sequence alignment (MSA), you should use the program hmmbuild:
# Build an HMM - note the unusual order with the output file first
hmmbuild my_hmm.hmm my_msa.fasta
# Search sequence(s) with an HMM
hmmsearch my_hmm.hmm my_sequences.fasta
So for annotation, we could take the Pfam family database and check our sequences against it for domains, and those can inform our functional annotation.
Exercise 5.3#
Exercise 5.3
Why might a protein database (Unitprot/Swissprot) be more advantageous than a nucleotide sequence database (GenBank/RefSeq, introduced in Sequence data) when exploring function?
Protein databases contain amino acid sequences with evidence of particular functions, whereas nucleotide sequence databases are focused on genome, gene and transcribed RNA sequences. Transcribed RNAs can also exist a regulatory RNAs that never encode for a protein.
Annotate the sequence mystery_sequence_03.fasta found in
/nfs/teaching/551-0132-00L/5_Annotationusing UniProt and Interpro web resources.
Firstly you will need to download the file to your computer using the scp command from your own machine (if you don’t remember how, go back to lesson 1 - Unix1 where we gave you an introduction to scp). Alternatively you can also open the file with less and copy/paste.
From the Uniprot website, you can perform a BLAST search (here) of the sequence against the Uniprot database. It should match a variety of XerC proteins from E. coli and Shigella.
From the InterPro website, you can perform a sequence search here but you will need to first convert the nucleotide sequence to protein sequence (for example using Python). You should find two domains in the protein, both integrases, though not exclusively phage-related as the sequence is from a bacteria.