Searching for alignments#
BLAST is used far more often for searching by alignment, that is, finding sequences in a database that align to a query (i.e. your sequence of interest). If we return to the BLAST homepage and again select Nucleotide BLAST (blastn), and then make sure that the “Align two or more sequences” is unchecked, we should see the standard search interface:
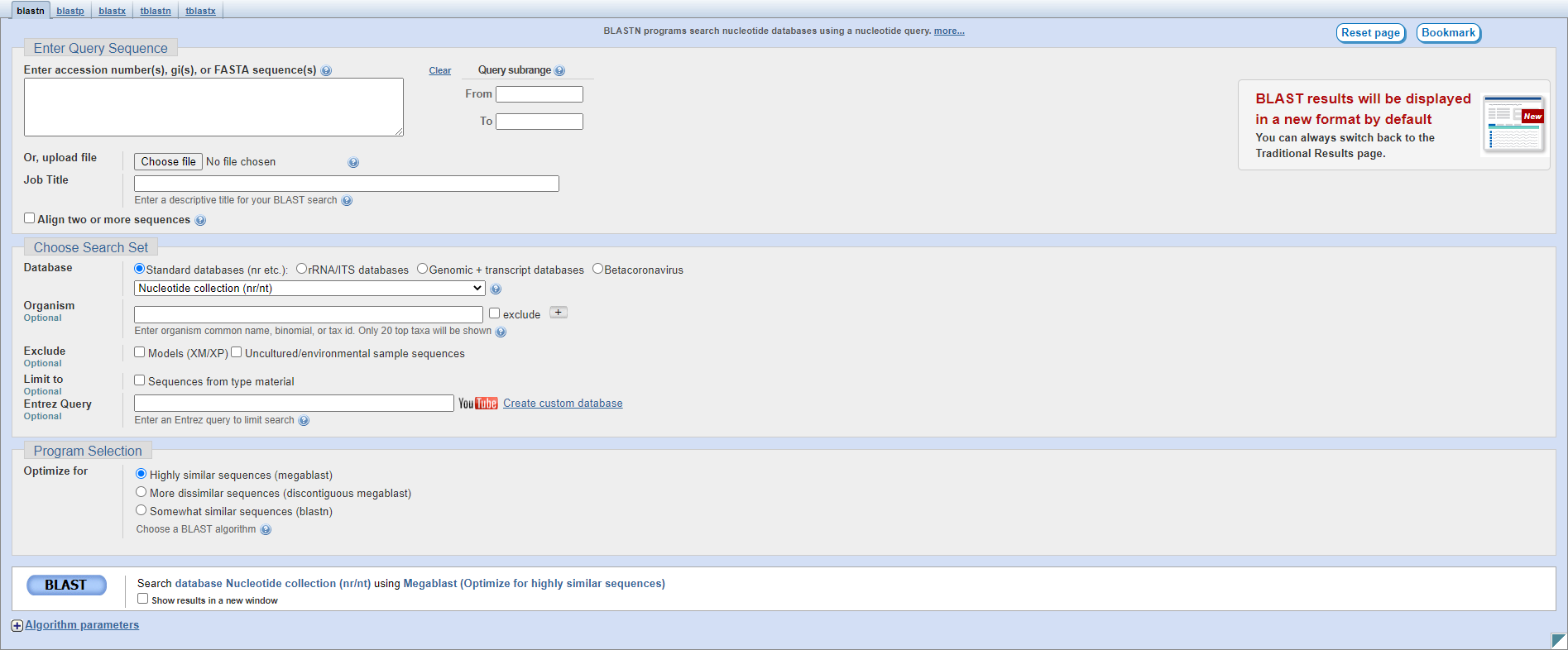
Let’s look at the different sections on this page:
Enter Query Sequence: this section is the same as for pairwise alignment.
Choose Search Set: here you can specify search parameters.
Database: this is where you select an NCBI database to search; typically nr/nt to search Genbank protein/nucleotide sequences but you may want to use RefSeq or another specific database.
Organism: you can enter the name of an organism or taxonomic level to restrict your search, for instance Escherichia or fungi.
Exclude: checkboxes to exclude RNA and protein sequences generated by NCBI’s automated pipeline (the Models), or uncultured sequences (those reconstructed from metagenomic samples).
Limit to: you can restrict your search to only type material, which are the exemplary species, see this paper for more information.
Entrez Query: you can also use custom search terms from the Entrez system to restrict your search.
Program Selection: you can select the specific algorithm you want to use here; the default is usually sufficient but you can use a slower but more sensitive algorithm if needed.
BLAST: this button will begin the search.
Algorithm parameters: here you can specify the exact parameters BLAST will use in alignment during the search; in general you won’t need to modify those used by the three Program Selection options.
Search results#
The results page has a summary of the search performed at the top (left), with the option to further filter the results (right).
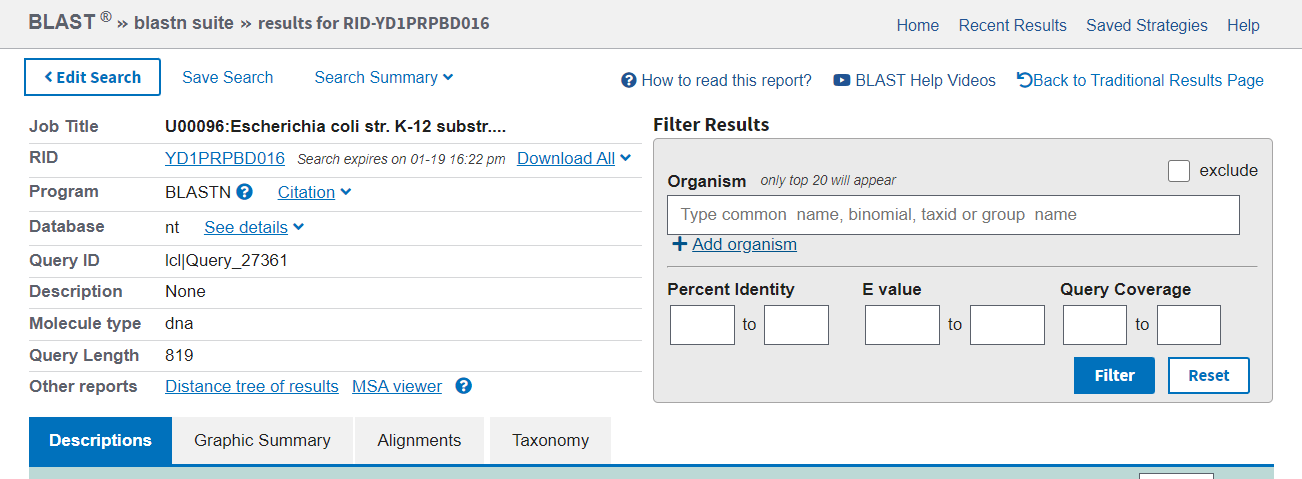
The first tab in the results section is Description. The statistics reported are the same as for pairwise alignment. You can sort hits by any of the data columns if you want to specifically find the longest or most identical hit, for instance, where the default is by e-value.
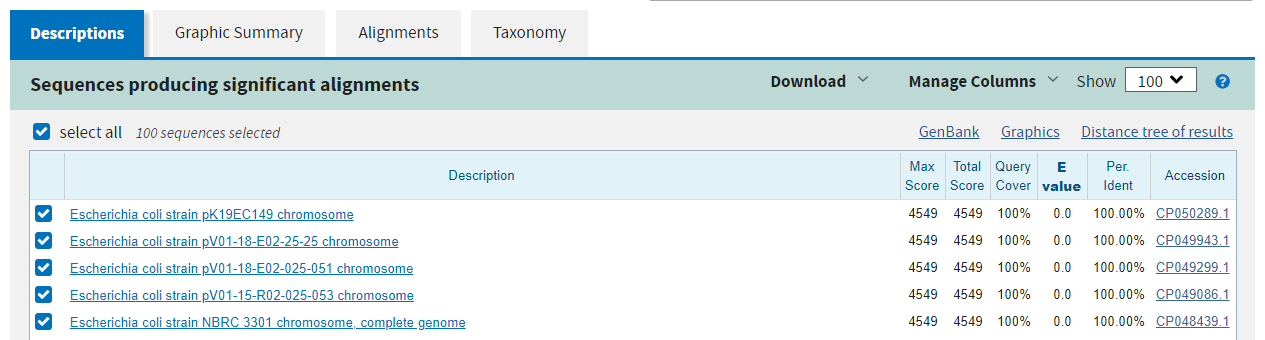
The second tab gives a graphical summary of the hits found. It depicts the quality of the alignment across the length of the query sequence with a colour code for alignment quality.

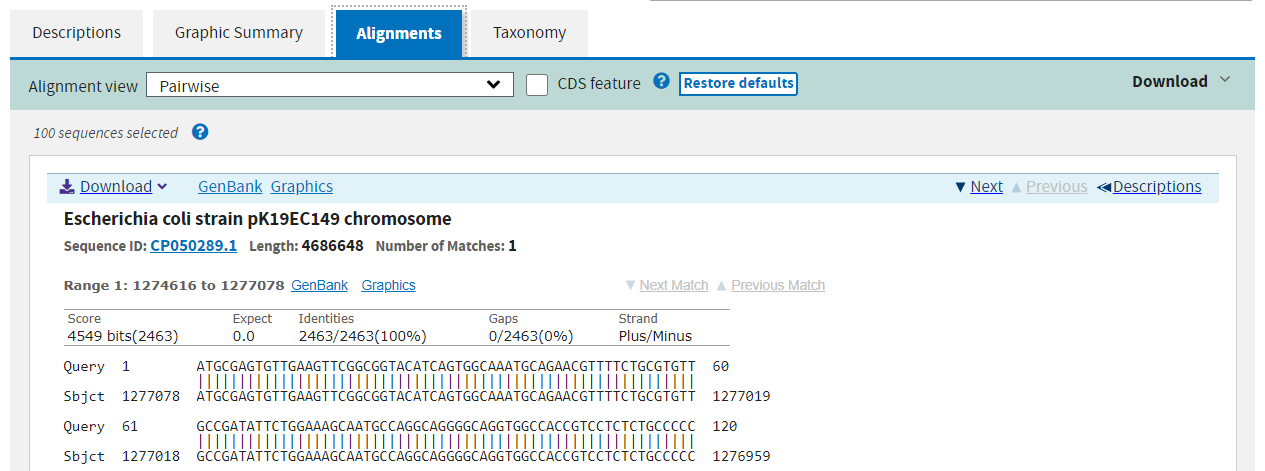
The third tab shows you the precise alignment found by the algorithm. The query and subject sequences are shown beside one another, with vertical pipe symbols “|” representing identity.
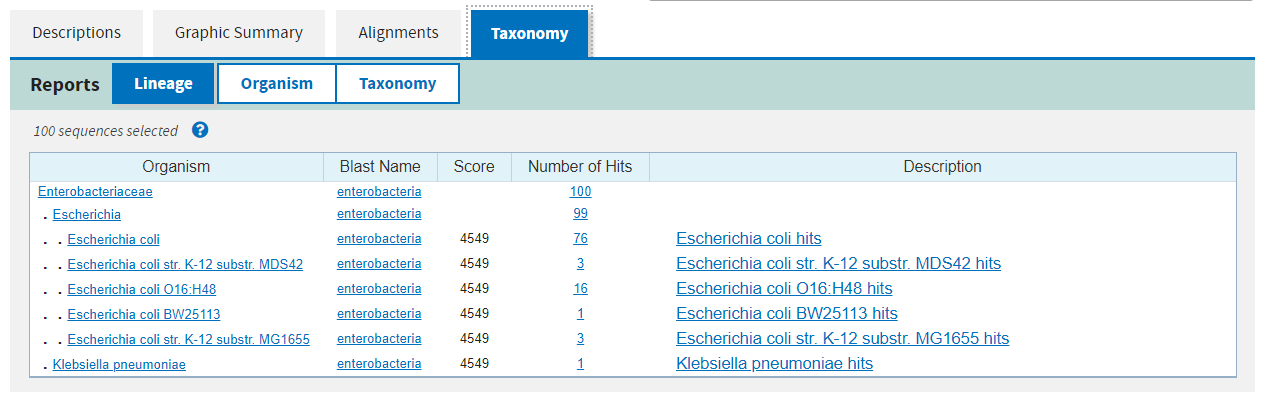
The fourth and final tab gives you a taxonomy of the organisms found in the hits and how many hits were found at each taxonomic level.
Exercise 4.3#
Exercise 4.3
Perform a nucleotide BLAST search for sequence found in the file
/nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_01.fasta
# Make sure the «Align two or more sequences» box is not ticked and the webpage has switched to the database search mode
# Open the mystery sequence and copy/paste it into the search field
less /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_01.fasta
Based on the results, what organism do you think the sequence is?
Based on the search you should find out that the sequence is from the Mycobacterium tuberculosis bacterium.
Based on that information, can you find a way to identify what protein the gene sequence encodes for? Three alternative solutions are mentioned below.
Go back to the NCBI blast starting page and select blastx (translated nucleotide to protein)
Make sure the «Align two or more sequences» box is not ticked and the webpage has switched to the database search mode
Enter your sequence or upload the file mystery_sequence_01.fasta as query
Note: Since multiple genetic codes exists, you ideally know at least what group of organisms a sequence belongs to before you try to investigate the protein it encodes, you can then decide which genetic code to use (in this case choose Bacteria and Archaea (11))
Note: You can also specify the organism (if known) to further increase the efficiency of your search
The result (mystery_sequence_03.faa) shows that the DNA sequence encodes for DNA topoisomerase (ATP-hydrolizing) subunit A/DNA gyrase subunit A from Mycobacterium tuberculosis
Go back to the NCBI blast starting page and select blastp (protein-protein alignment)
Make sure the «Align two or more sequences» box is ticked
Copy/paste the protein sequence from the previous exercise in the first entry (click on Alignments subpage, click on first alignment, click on the sequence ID, click on FASTA, copy/paste) or copy/paste or upload from file mystery_sequence_03.faa
Open the file mystery_sequence_04.faa and copy/paste this protein sequence into the second entry (or upload the file)
Checking the alignment, you will see that there are 62 positions with differences in encoded amino acids
If you are instead interested in the total number of differences between the two genes encoding for the two proteins, you would want to compare them at the DNA level. Why is this the case and how would you do this?
Because of the redundancy of the genetic code, some triplets of DNA nucleobases encode for the same amino acid. Thus differences in DNA sequences leading to the same amino acid are not visible at the protein level.
Make sure the «Align two or more sequences» box is not ticked and the webpage has switched to the database search mode
Go back to the NCBI blast starting page and select tblastn (protein to translated nucleotide)
Enter the mystery_sequence_04.faa and retrieve the DNA sequence from the results
Note that the output of this search returns the complete genome sequence instead of only the gene (this does not matter much though because BLAST is a local alignment which will still find alignments also between sequences of very different length where most parts do not align)
Go back to the NCBI blast starting page and select blastn (nucleotide-nucleotide alignment)
Make sure the «Align two or more sequences» box is ticked
Enter the mystery_sequence_01.fasta as first entry
Enter the DNA sequence that you just translated from mystery_sequence_04.faa as a second entry (mystery_sequence_05.fasta)
Checking the alignment, you will see that there are 340 positions with differences in the DNA sequence
Note that the 62 amino acids could only lead to a maximum of 3*62=186 changes in amino acids, thus the rest of the differences are only present at the DNA level but not at the protein level