Prediction of the impact of protein mutations using FoldX#
The FoldX algorithm (https://foldxsuite.crg.eu) can be used to estimate both the stability of a protein (𝚫G) and its change upon mutation (𝚫𝚫G). It relies on statistical energy functions that have information on different properties of bonds, clashes, entropy, etc. It has a manual with information on the different capabilities and functions. To test FoldX we will be using the same structure of the human protein kinase A (5J5X). As FoldX generates several files during the process it will be best to create a dedicated directory to use it. In the terminal we can make a new directory, navigate to it and download the PDB file. To run FoldX you need to access the terminal, either by using SSH or by going to the terminal window in Rstudio. In the terminal we can make a new directory, navigate to it and download the PDB file. To use FoldX we will first have to import the FoldX module.
mkdir foldxtest
cd foldxtest
wget https://files.rcsb.org/download/5J5X.pdb
ml FoldX
FoldX requires a file “rotabase.txt” in the local directory where it is being used. This can be copied from a location within the same server:
cp /nfs/nas22/fs2202/biol_micro_teaching/software/easybuild/software/FoldX/4.0/bin/rotabase.txt ./
In order to first use FoldX to estimate the impact of mutation we must first attempt to repair it to the most stable state as predicted by FoldX. Running the RepairPDB command, will ask FoldX to change the conformation of the side-chains to avoid clashes and optimize overall the stability of the structure. FoldX does change the backbone of the structure, only moving the side-chains. This repair can take 5-10 minutes to conclude depending on the size of the protein.
foldx --command=RepairPDB --pdb=5J5X.pdb
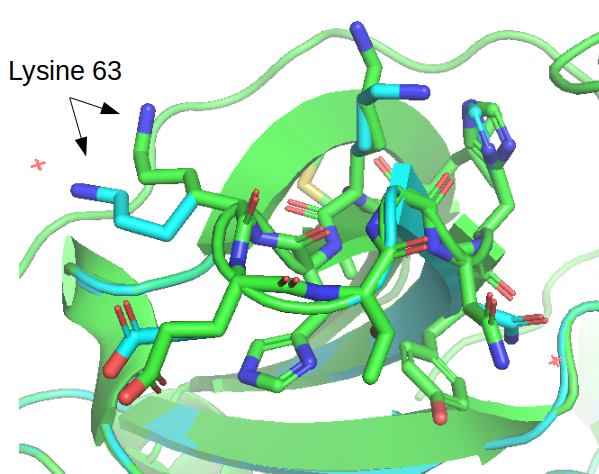
After running this the estimated stability of the protein 𝚫G went from +82.95 kcal/mol to –55.43 kcal/mol. There is a detailed description of the many small changes that led to the very large difference in predicted stability. The repaired structure is outputted in the same folder and the figure below illustrates a few cases where the conformations of the side changes were moved between the original structure and the repaired structure.
You can retrieve the repaired PDB to your local computer via the export function in Rstudio or using the scp command:
scp <user>@cousteau.ethz.ch:./foldxtest/5J5X_Repair.pdb ./
Generating mutated structures and estimating the impact on the stability can be done using the BuildModel command. It requires a text file holding a list of mutations to test with each mutation in a line in the format such as “LA224A;” corresponding to starting amino acid (L), the chain (A), the position (224) and the amino acid we want to mutate it to (A). As an example we can attempt to predict the impact of mutations in two positions, L at position 224 that is part of the core of the protein and I at position 339 that is at the surface. In R studio workbench we create a new text file and write in the following mutations:
LA224A;
LA224E;
LA224W;
IA339A;
IA339E;
IA339W;
Then save this file as “individual_list.txt” in the same foldxtest directory. Back in the terminal the prediction of the impact of mutations can be predicted using the following command:
foldx --command=BuildModel --pdb=5J5X_Repair.pdb --mutant-file=individual_list.txt
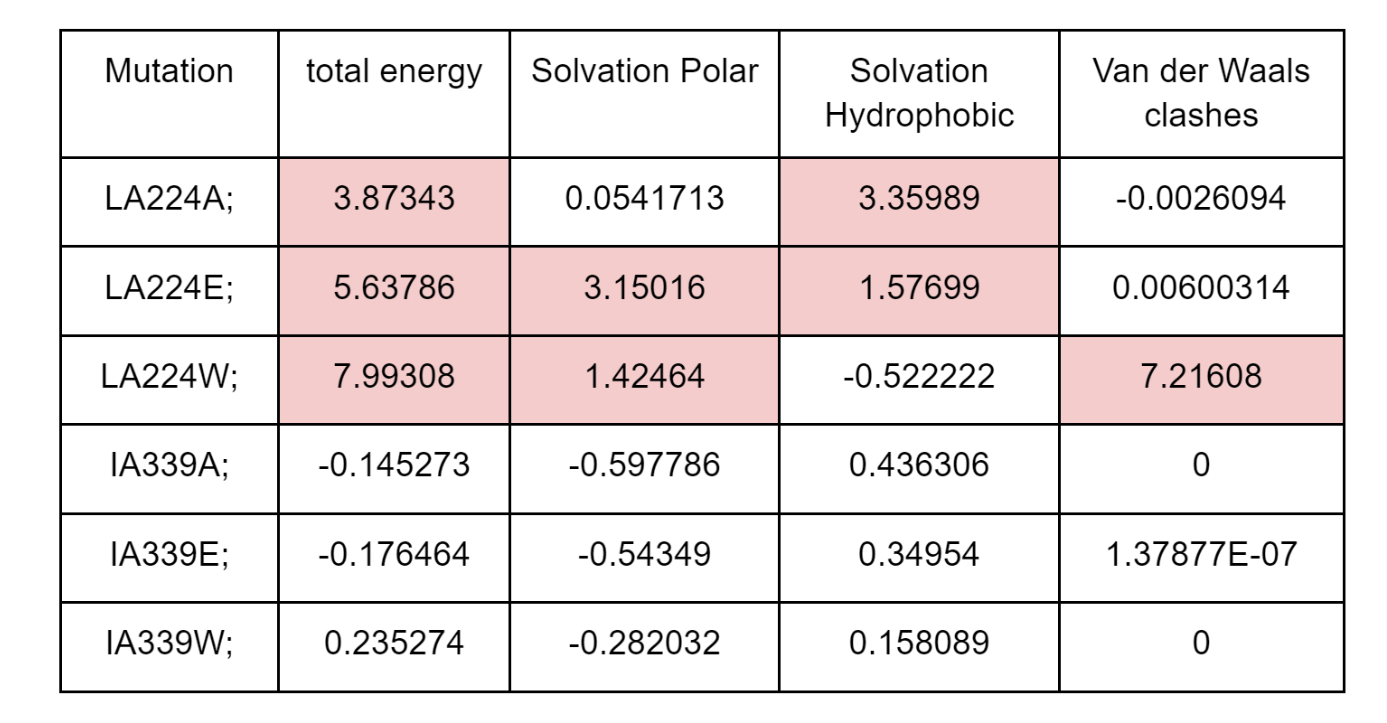
FoldX will generate an output PDB file with each of the mutated structures and the summary of predicted energy differences in “Dif_5j5x_Repair.fxout” which will have an entry per mutation in the same order as the list of mutations. The output contains information on the total 𝚫𝚫G as well as different components that contribute to the total score. As shown in the table below, the 3 mutations in the L224 core residue are predicted to be destabilizing with 𝚫𝚫G>2 kcal/mol. However, there are different reasons for the detrimental effect, with the mutation to the large tryptophan (W) causing a strong clash and the mutation to the small alaline (A) having a defect in solvation hydrophobicity, likely due to leaving a “hole” in the core of the structure. The mutation to the negative charge glutamate residue (E) causes issues with having hydrophilic charged residues in the core of the protein (solvation polar energy). The corresponding mutation in the surface residue I339 has essentially no predicted effect on protein stability.
In addition to BuildModel, FoldX can also perform a fast calculation of the impact of the mutation of every single residue to alanine. This can be achieved using the “AlaScan” command which is faster but less accurate than estimates obtained using the BuildModel command.
foldx --command=AlaScan --pdb=5J5X_Repair.pdb
The output of this command is a file “5j5x_Repair_AS.fxout” containing the predicted impact of mutating each residue to alanine in seperate lines. For example: GLY 9 to ALA energy change is 0.801459
Fold it#
We are finished this week with Structural Bioinformatics and as such there is no homework for this week. Instead, you are encouraged to play the FoldIt g ame. This game teaches the gamer how to fold proteins in a visual way. It is also used for deriving actual protein structure predict ions by aggregating the accumulated experience of the best folders. For example, FoldIt players have successfully predicted the structure of an HIV protei n and have been acknowledged for this in the author list of the paper.