Using BLAST online#
Most researchers are familiar with the web-based interface for BLAST found here.
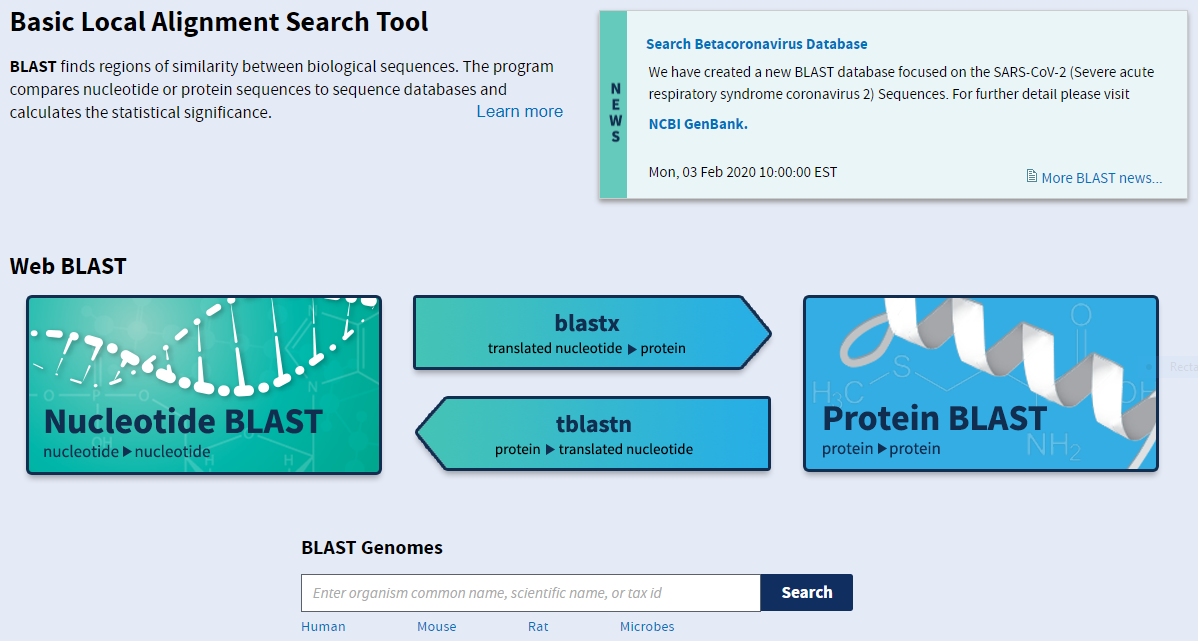
The four main varieties of BLAST are neatly summarised on this frontpage.
blastn: nucleotide blast for comparing DNA sequences (left)
blastx: searching a protein database for a translated nucleotide query (middle top)
tblastn: searching a translated nucleotide database for a protein query (middle bottom)
blastp: protein blast for comparing protein sequences (right)
You should therefore choose your BLAST algorithm based on the nature, nucleotide or protein, of your query and the database against which you want to compare it. There are also further versions available for more specialised applications.
More details about BLAST on the NCBI-Webpage can be found here.
Pairwise alignment#
We will first select Nucleotide BLAST (blastn) to perform a pairwise alignment of two nucleotide sequences that we have put in the files /nfs/teaching/551-0132-00L/4_Alignment/pairwise1.fasta and /nfs/teaching/551-0132-00L/4_Alignment/pairwise2.fasta.
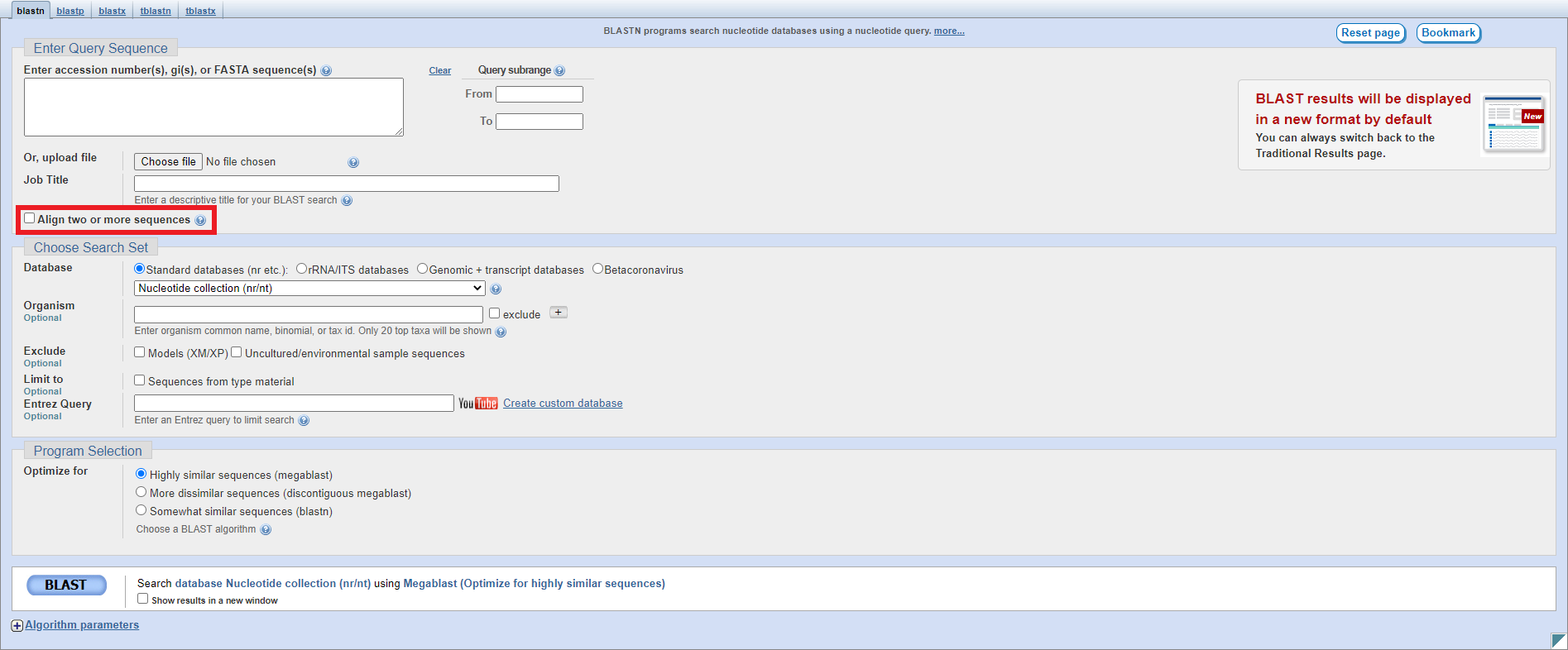
The setup page for blastn looks as follows - you should click on the checkbox highlighted to enable pairwise alignment:
This will change the page to look as follows:
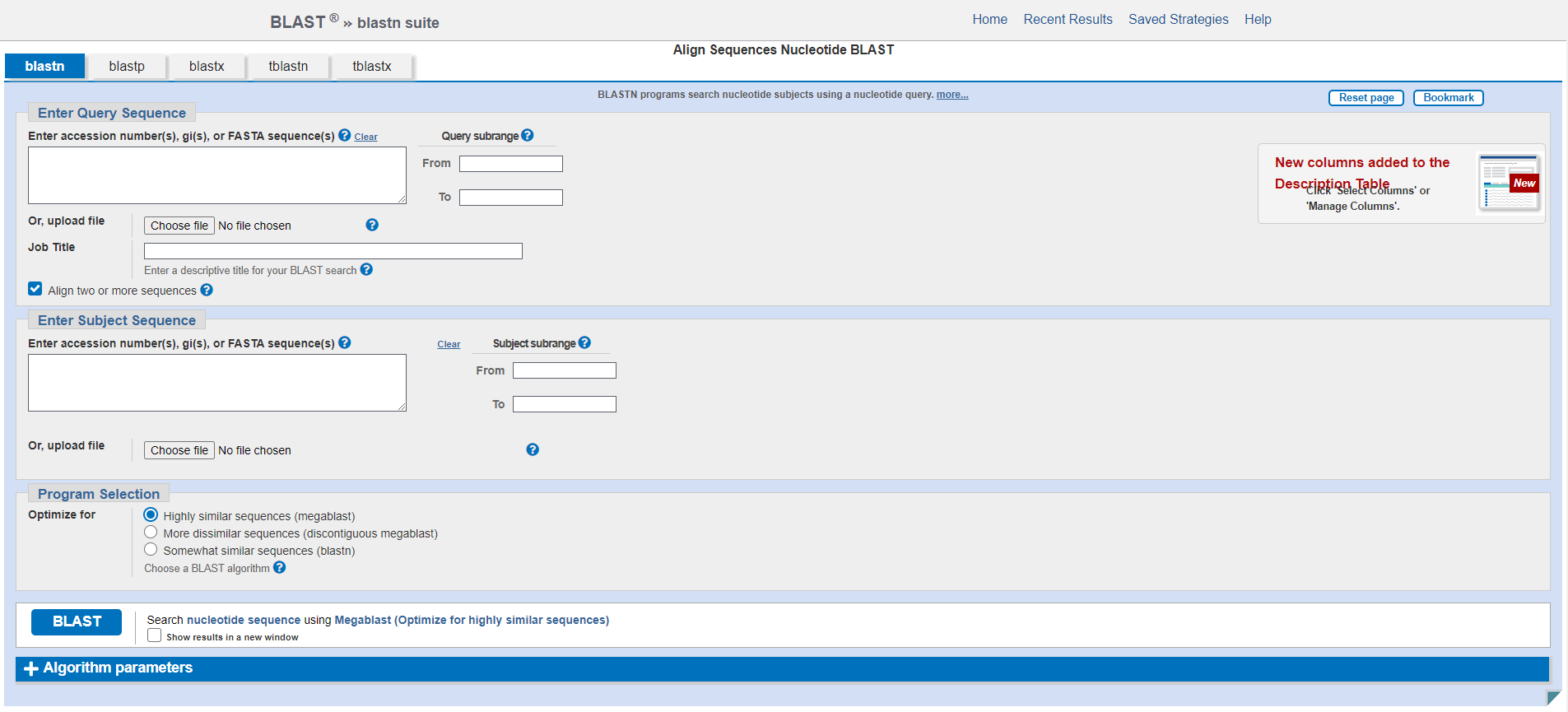
In each of the sections Enter Query Sequence and Enter Subject Sequence you can either paste the relevant sequence into the text box directly or choose a file to upload, which should be in fasta format. For each sequence you can also specify a subrange from the sequence by giving start and end coordinates. You can also give your alignment a Job Title.
The Program Selection section allows you to select which specific blastn algorithm you want to use. Depending on your application and more specifically the expected sequence similarity you may want to choose a different program to optimize your search and algorithm speed. The default program megablast is very fast and works best for highly similar sequences (with expected sequence percent identity >= 95%). If you expect your sequences to be a bit more dissimilar, you might want to use discontiguous blast which allows for more mismatches. blastn is the slowest alorithm but allows much shorter alignments and therefore enables to retrieve alignments between somewhat similar (more distant) sequences.
Exercise 4.1#
Exercise 4.1
Perform a pairwise nucleotide BLAST alignment of the sequences in the files
/nfs/teaching/551-0132-00L/4_Alignment/pairwise1.fastaand/nfs/teaching/551-0132-00L/4_Alignment/pairwise2.fasta. For this example, you don’t need to enter any subrange coordinates or change the algorithm from the default (megablast). It is up to you whether you want to copy and paste the sequences or upload the fasta files.
# Make sure the "Align two or more sequences" box is ticked
# Open the file pairwise1.fasta with less and copy the nucleotide sequence and paste under "Enter Query Sequence"
less /nfs/teaching/551-0132-00L/4_Alignment/pairwise1.fasta
# Repeat for pairwise2.fasta but paste the sequence under "Enter Subject Sequence"
less /nfs/teaching/551-0132-00L/4_Alignment/pairwise2.fasta
# You can also download the files first with the scp command you learned in Unix1 and upload the files to NCBI
As we learned previously, megablast works well for sequences which are highly similar and is very fast, so in this case we can use megablast because we know that the two example sequences are highly similar
Investigate the result by reading through the “Descriptions” tab, specifically the parameters Max Score, Query Coverage and Percent Identity
Check the alignment in detail by clicking on the tab “Alignments”
Alignment results#
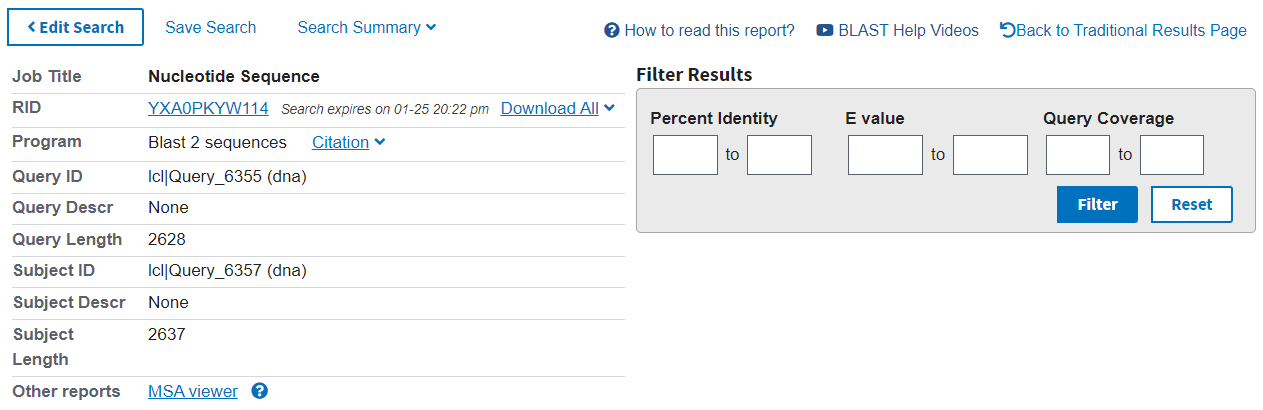
The results page has a summary of the search performed at the top (left), with the option to further filter the results (right).
The first tab in the results section is Description. Here you can see the statistics for each alignment found between the two sequences:
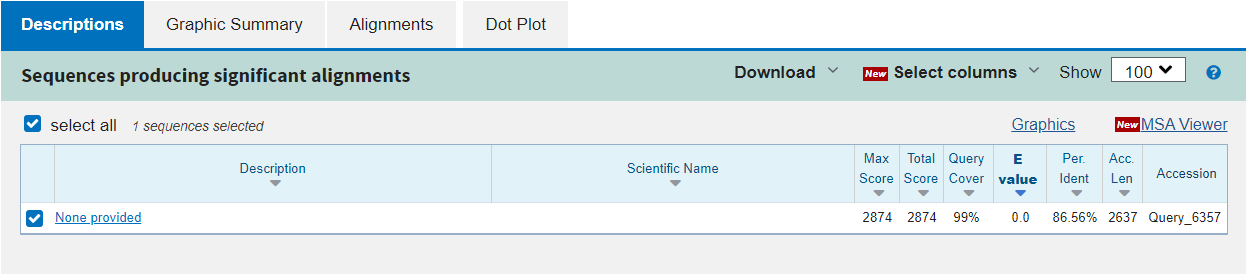
Max Score: the highest score from the alignments found
Total Score: the sum of scores for all alignments found
Query Cover: the percentage of the query sequence for which any alignment was found
E value: the likelihood of the alignment being seen by chance (note that this is dependent on the database size searched)
Per. Ident: the percentage identity of the alignment, i.e.: how many base pairs are identical
Acc. Len: the length of the subject sequence
Accession: the name of the subject sequence (or an arbitrary name)
In this example, there is only one alignment in the results and so some of this information is not interesting. What we can see is that the alignment covers 99% of our query sequence and is approximately 87% identical. We cannot really say if this alignment is significant or not because we have only compared our query to one subject, and this was contrived to give a successful alignment. Nonetheless we can inspect the alignment more carefully in the other tabs.
Warning
The ‘percent identity’ of an alignment is sometimes, perhaps misleadingly, referred to as ‘percent homology’ - however, this implies a shared evolutionary history. As two sequences that perform the same function could well have evolved independently, sequence similarity suggests but is not sufficient proof of homology.
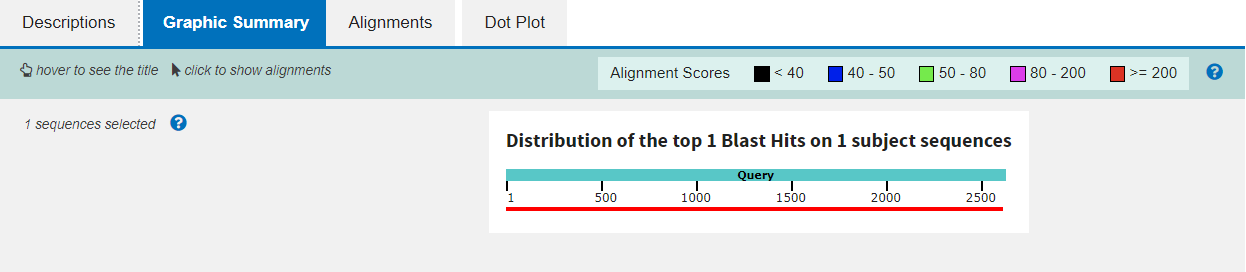
The second tab gives a graphical summary of the alignment, depicting the quality across the length of the query sequence with a colour code:
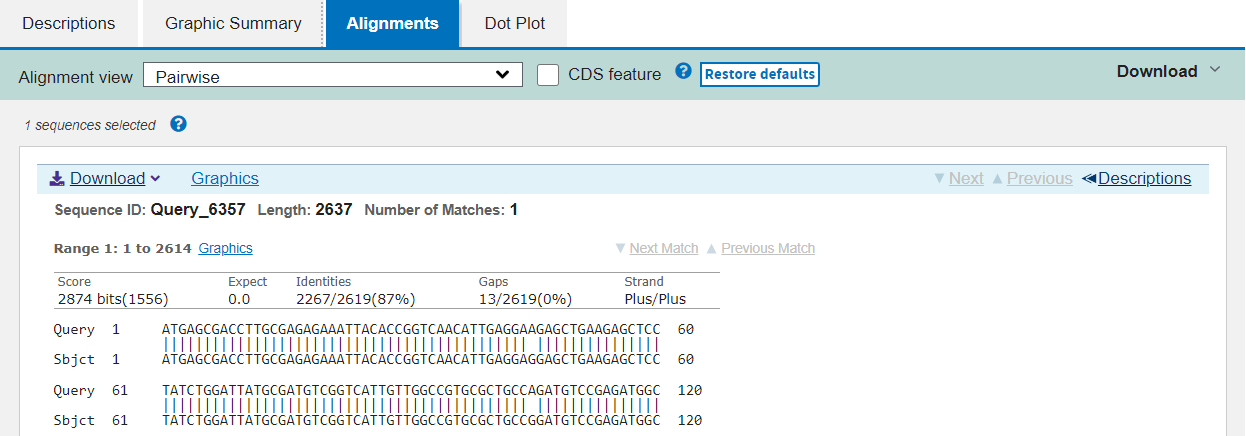
The third tab shows you the precise alignment summarised in the first tab. The query and subject sequences are shown beside one another, with vertical pipe symbols “|” representing identity and dashes “-” for gaps in either sequence.
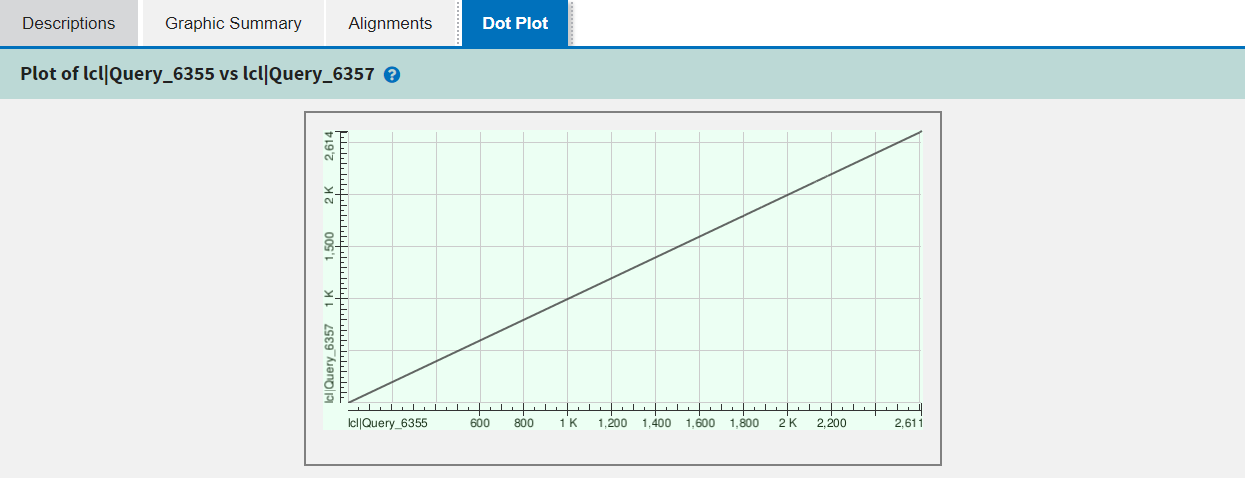
The fourth and final tab is specific to pairwise alignment and shows a dot plot of the alignment or alignments between the pair of sequences.
Exercise 4.2#
Exercise 4.2
For the alignment you just performed, what would happen if you enter subrange coordinates from 1 to 600 for pairwise1.fasta and from 500 to 1000 for pairwise2.fasta? You might have an idea based on your previous result, but if you are not sure you can simply try it out.
Repeat the alignment with 100 bases overlapping subrange coordinates for the two sequences
You will see that there is no longer an alignment between the two sequences even if we know that they align along the whole sequence length and also along the subrange overlap
This is important to realize because subrange coordinates should be used with care and only if 1) you are certain that there is no similarity in the disregarded ranges or 2) you are only interested in alignments within a certain range
What would happen if you extend the subrange coordinates from 1 to 750 for pairwise1.fasta and you keep the subrange from 500 to 1000 for pairwise2.fasta? If you are not sure you can simply try it out.
Repeat the alignment with a bigger overlap between subrange coordinates for the two sequences
Extending the subrange overlap allows you to recover the sequence alignment
Depending on the number of mismatches and gaps the total alignment score will be impacted so that the sequence alignment needs to be a certain length to show up as alignment
You can check the tab “Alignments” again and will see that only the subrange overlap shows up because BLAST is a local alignment algorithm and thus the rest of the sequences that does not align is ignored
Can you think of a different way to retrieve the alignment between the subrange 1 to 600 for pairwise1.fasta and the subrange 500 to 1000 for pairwise2.fasta? Hint: If we do not change the chosen coordinate subranges and sequences, the retrieval of the alignment will be directly dependent on the scores/penalties of matches/mismatches/gaps defined by the alignment algorithm.
As we learned previously, megablast works well for sequences which are highly similar and is very fast
If we do not know how similar/distant two sequences are, we would rather use the discontiguous megablast or blastn algorithm which are slower but can retrieve more distantly related sequences
If we select either of the slower algorithms we can retrieve the alignment of 100 bases alignment between the two sequences that we could not retrieve with megablast
However is important to note that these other algorithms are much slower and take longer until they display a results