Sequence data#
DNA and protein sequences are fundamental to our understanding of biology and with the advent of high-throughput sequencing technologies, we generate ever more sequencing data at an ever increasing rate. Sequence data is nowadays involved in almost every biological project in one form or another and it is consequently very likely that you will encounter and work with sequence data if you stay in biology (designing a primer, working with CRISPR or checking the genotype of a mouse; all these tasks use sequence data even though they are not directly linked to bioinformatics). Therefore, it is crucial to understand this type of data.
At the core of our ability to use this data are two standard file formats:
FASTQ - this is data from DNA sequencing that includes a quality score for each base
FASTA - this is processed data, and can be DNA or protein sequence
FASTQ format#
A fastq file may contain multiple sequences in fastq format. This is a text-based format where each entry consists of 4 lines:
The sequence identifier, probably automatically generated by the sequencing device; the line always begins with @
The raw sequence, consisting of A, C, G, T and perhaps N (standing for bases that cannot be clearly assigned)
These days unused, but in theory a repeat of line 1; the line always begins with +
The quality score for each base in the raw sequence
@071112_SLXA-EAS1_s_7:5:1:817:345
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
Quality scores#
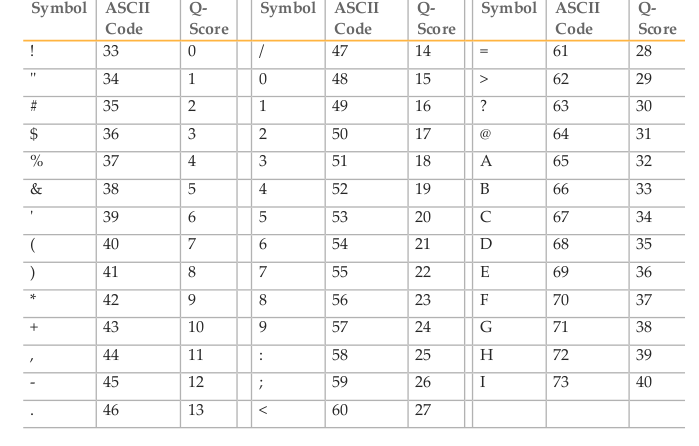
Quality scores in the standard Phred format range between 0 and 93, although sequencing read data is rarely higher than 60. The scores are encoded such that there is one character per base in the 2nd line of the fastq entry, using the ASCII encoding table (a computing standard). For instance, the letter A has a decimal equivalent of 65. Sanger format offsets the quality score by 33, so this represents a quality score of 32 (65-33).
The Phred quality score (Q) is logarithmically (base 10) related to the error probability (E) and can therefore be interpreted as an estimate of error (E) or as an estimate of accuracy (A).
This formula gives us the following benchmarks for accuracy:
Phred Quality Score |
Error |
Accuracy |
|
|---|---|---|---|
10 |
10% |
90% |
|
20 |
1% |
99% |
|
30 |
0.1% |
99.9% |
|
40 |
0.01% |
99.99% |
|
FASTA format#
A fasta file may also contain multiple sequences in fasta format (sometimes known as multi-fasta). The sequences will be either DNA or protein sequences, but never mixed in the same file. The fasta format is text-based, where each sequence entry consists of 2 elements:
A header line beginning with the > character and some information about the sequence such as ID, species name, gene name or position
One or more lines of sequence data. When containing multiple lines, each line is usually 70 or 80 characters long, and this is done only for readability.
# FASTA format of nucleotide sequence
>Mus_musculus_tRNA-Ala-AGC-1-1 (chr13.trna34-AlaAGC)
GGGGGTGTAGCTCAGTGGTAGAGCGCGTGCTTAGCATGCACGAGGCCCTGGGTTCGATCC
CCAGCACCTCCAGGG
# FASTA format of protein sequence
>gi|5524211|gb|AAD44166.1| cytochrome b [Elephas maximus maximus]
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLV
EWIWGGFSVDKATLNRFFAFHFILPFTMVALAGVHLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLG
LLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIVIL
GLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGX
IENY
Conditional pattern searches#
To extract information from sequence data files, it may be helpful to understand the concept of regular expressions, described in detail in the “Additional concent” in the section “Introduction to Unix”. Regular expressions allow you to make your pattern searches with grep more complex through adding restrictions, conditions or character groups rather than just searching for a single word. Here we want to introduce two important conditional regular expressions to you:
^ stands for the start of a line, grep “^pattern” will search for “pattern” occurring at the start of a line.
$ stands for the end of a line, grep “pattern$” will search for “pattern” occurring at the end of a line.
# FASTA file mus_musculus_trna.fasta with nucleotide sequence
>Mus_musculus_tRNA-Ala-AGC-1-1 (chr13.trna34-AlaAGC)
GGGGGTGTAGCTCAGTGGTAGAGCGCGTGCTTAGCATGCACGAGGCCCTGGGTTCGATCC
CCAGCACCTCCAGGG
# Search for lines containing "GGG"
grep "GGG" mus_musculus_trna.fasta # There are multiple occurrences
# Search for lines containing "GGG" at the start of a line
grep "^GGG" mus_musculus_trna.fasta # There is 1 occurrence at the start
# Search for lines containing "GGG" at the end of a line
grep "GGG$" mus_musculus_trna.fasta # There is 1 occurrence at the end
Exercise 3.1#
Exercise 3.1
/nfs/teaching/551-0132-00L/3_Sequence/ contains example files example_reads_R1.fastq, example_reads_R2.fastq and example_sequences.fasta.How might you count the number of entries (sequences) in a multi-fasta file using command line tools? HINT: Think carefully about the ways your method might go wrong, consider using a regular expression.
# Enter directory
cd /nfs/teaching/551-0132-00L/3_Sequence/
# Count fasta records
grep -c "^>" example_sequences.fasta # count lines starting with ">" within fasta file
# Description of command
# grep "", searches for regular expressions within quotation marks ""
# -c (option), prints the total count of occurences of the searched pattern instead of selected lines
# ^, means 'start of line'
# "^>", searches for lines starting with >
# example_sequences.fasta (argument), the file that the command should be executed on
How about for a fastq file? HINT: Think carefully about the ways your method might go wrong, consider using a regular expression.
# Count fastq records
grep -c "^+$" example_reads_R1.fastq # count lines that contain only one "+" from start to end of line within fastq file (3rd line)
# Description of command
# grep -c "" # count occurence of pattern within quotation marks ""
# ^ # means 'start of line'
# $ # means 'end of line'
# "^+$", searches for lines that only contain one "+" between the start "^" and end "$" of a line
How can you answer the question above with commands used for data wrangling instead of pattern searches?
# Count numbers of lines and extract the result from your output
wc -l example_reads_R1.fastq | cut -d " " -f 1
# Description of command
# wc -l example_reads_R1.fastq # prints total number of lines in file and filename (separated by whitespace, because can be used on multiple files)
# \| # pipe is used as connector between two commands so that second command can be executed on output of first command directly
# \| cut -d " " -f 1 # uses output of previous command, separates output by whitespace and prints only first field/column (so only number of lines, without filename)
# Number of lines across all entries
4000 # Remember that you have to divide this number by 4, as each entry in a fastq file contains 4 lines
# Number of entries
1000
In the example fastq entry shown above
example_reads_R1.fastq, calculate the Phred quality scores for the final 4 bases.
# Quality score calculation
# Final 4 bases have the symbols 4, %, ), @
# In ASCII code that's: 52, 37, 41, 64
# In Phred (ASCII-33) that's: 19, 4, 8, 31