Predicting protein structures using AlphaFold2#
AlphaFold can be easily used to make novel protein structure predictions for your sequence of interest. This can be done via the browser using an easy to use colab notebook called ColabFold.
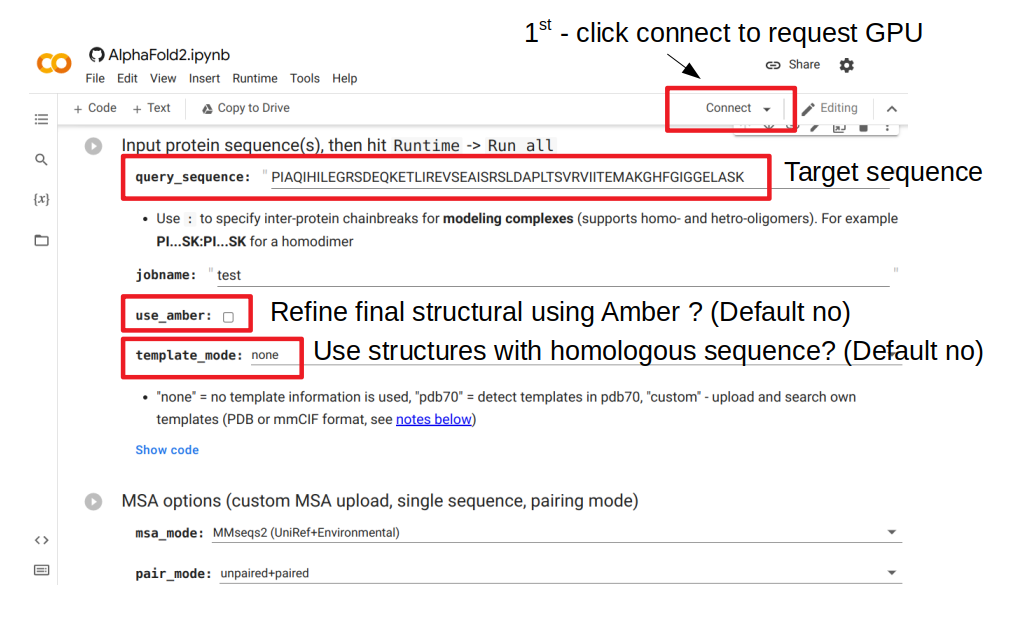
The figure above shows the important parameters to consider when making a prediction using this web server. The first thing to do is to click connect, which will request a GPU node to serve as resources for this run. The query_sequence element needs to be changed to the target sequence to model. Then the user needs to decide if they want to refine the structure after it is predicted; this will attempt to finetune some of the details to improve bonds and avoid clashes. This additional step is slow and often not very useful. The user can also choose to use or not structures from sequences that are homologous to the target sequence (“template_mode”). By default it is set to “none” which means no use of template. Changing this value to “pdb70” will add a step where homologous structural templates are used to further improve the result.
After all selections were made, the prediction can be generated by going to the menu and selecting Runtime -> Run all. This will take some time that depends on the size of the protein and will generate a predicted structure and some useful plots that relate to the confidence in the prediction.
Predicting the impact of protein mutations#
Mutations can be introduced in DNA during the normal functioning of any cell. Some of these changes can lead to diseases such as cancer but they are also the source material for selection to act upon during the course of evolution. There are many types of genetic changes such as large deletions or insertions or smaller single nucleotide changes. When these changes occur within protein coding regions they have the potential to change the coding amino-acids and consequently also the structure and function of proteins. Given the importance of such changes, there has been a large number of methods developed to predict when such mutations have an impact on protein function. There have been two major types of predictors, ones that rely primarily on information from sequence evolution and others that rely primarily on protein structure.
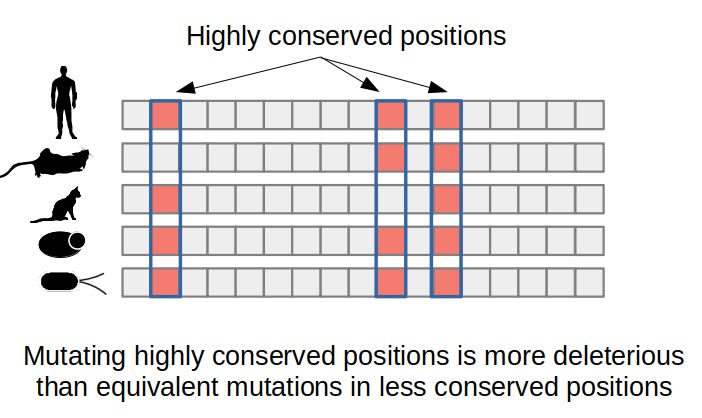
Predicting the impact of a mutation from evolution related information primarily makes use of multiple sequence alignments of the protein of interest together with homologous sequences, usually orthologs (i.e. same protein in different species). Analyzing the resulting alignment can identify which amino-acid changes occur less often in natural sequences and therefore are likely to be deleterious to protein function. For example, enzyme catalytic residues are often extremely conserved and this conservation can be easily detected in a multiple sequence alignment. The SIFT algorithm is an example of a computational method that uses protein sequence conservation to predict the impact of protein mutations.