Additional content#
This content is an additional resource for interested students, who want to learn about a wider variety of topics and commands used by scientists working on the command line on a daily basis.
File name conventions#
In Unix systems there are only really two types of files: text or binary. The file name ending (.txt or .jpg) doesn’t really matter like it does in Windows or Mac OS, however it is used to indicate the file type by convention. Some file types you will encounter include:
.txt - A generic text file.
.csv - A ‘comma separated values’ file, which is usually a table of data with each line a row and each column separated by a comma.
.tsv - A ‘tab separated values’ file, which is the same but separated by tab characters.
.fasta or .fa - A fasta formatted sequence file, in which each sequence has a header line starting with ‘>’.
.fna - A fasta formatted nucleotide sequence file, usually gene sequences.
.faa - A fasta formatted protein sequence file.
.sh - A ‘shell script’, which contains commands to run.
.r - An R script, which contains R commands to run.
.py - A python script, which contains python commands to run.
.gz or .tar.gz - A file that has been compressed using a protocol called ‘gzip’ so that it takes up less space on the disk and transfers over the internet faster.
Wildcards#
When providing a file path as an argument to a command, it is often possible to provide multiple file paths using wildcards. These are special characters or strings that can be substituted for a matching pattern. For many commands using wildcards allows you to execute the associated action on each file that matches the pattern, though this obviously does not work in all cases.
‘?’ matches any single character
* matches any number of any characters
‘[…]’ matches any character within the brackets
‘{word1,word2,…}’ matches any string inside the brackets
For instance:
# Pattern matching
ls /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/* # lists all files in the ecoli directory
ls /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/*.fna # lists all nucleotide fasta files there
ls /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/*.f?a # lists all nucleotide and protein fasta files there
Searching for a file#
When you are trying to find a file in your system, the command find offers a number of options to help you. The first argument is where to start looking (it looks recursively inside all directories from there), and then an option must be given to specify the search criteria.
# Finding files ("." stands for the current directory you are in)
find . -name "*.txt" -type f # searches for files ending in .txt. The type option defines the type of the file.
find . -mtime -2 # searches for files modified in the last two days
find . -mtime +365 # searches for files modified at least one year ago
find . -size +1G # searches for files at least 1GB or larger
find . -maxdepth 1 # searches only on one level, thus only here, i.e.: doesn't look inside directories
Advanced Exercise A1.1#
Advanced Exercise A1.1
Navigate to the
/nfs/teaching/551-0132-00L/1_Unix/genomes directory
# Navigation
cd /nfs/teaching/551-0132-00L/1_Unix/genomes
Use man to read about the find function
#Looking at find
man find
Use find to get a list from everything stored in the
/nfs/teaching/551-0132-00L/1_Unix/genomes directory
# Getting a list with find
find /nfs/teaching/551-0132-00L/1_Unix/genomes/
Use find to look for all .faa files there
# Looking fore .faa files
find . -name "*.faa"
Use find to look for all files larger than 5MB
# Looking fore files lager than 5MB
find . -size +5M
Now combine these criteria to find all .faa files larger than 5MB
# Looking fore .faa files larger than 5MB
find . -name "*.faa" -size +5M
Regular Expressions#
The reason this happens is that in the context of these search functions, “.” represents any character. It is acting as a wildcard, from a different set of wildcards to those discussed in Unix1.
This set of wildcards is part of a system of defining a search pattern called regular expression or regex. Such a pattern can consist of wildcards, groups and quantifiers, and may involve some complex logic which we will not cover here. Further, the exact set of wildcards available depends on the programming language being used.
# Wildcards and quantifiers
. any character
\d any digit
\w any letter or digit
\s any whitespace
^ the start of the string
$ the end of the string
* pattern is seen 0 or more times
+ pattern is seen 1 or more times
? pattern is seen 0 or 1 times
These are just a few of the possibilities available. An example regular expression that would search for email addresses, for instance, would be:
# name@domain.net can be matched as: \w+@\w+\.\w+
Let’s break this down:
The first part
\w+looks for any letter or digits one or more times, i.e.: the name part of the email address. Note that w does not match punctuation like “.” but does match underscores “_”.Then we ask for an at symbol
@.The second part
\w+again matches any alphanumeric string, i.e.: the domain part of the email address.Then we ask for an explicit full stop
\.which has to be delimited because a normal “.” matches any character.The third and final part is the same as the first and second and should match the net part of the email address.
So this is not a perfect regex for all email addresses because they can contain full stops and have more complex domain addresses.
Instead of searching for a regular expression describing a class such as w’ standing for any letter or digit, you could search for a specific expression such as the sequence ‘ACGT’. Enclosing this expression in brackets ‘()’ turns it into a group. You can then also search for multiple occurences of this group by using brackets ‘()+’ or you can simultaneously search for multiple patterns using the pipe character ‘|’. The pipe character acts as a logical OR operation here and divides the individual expressions within the group into alternates.
# Multiple occurences
ACGT would match ACGT
(ACGT)+ would match ACGT, ACGTACGT, ACGTACGTACGT etc.
(AC|CG|GT) would match AC, CG, GT
Advanced data wrangling#
In this section, we cover some additional data wrangling commands which are commonly used by people working with large amounts of data.
The command cut allows you to extract a single column of data from a file, for instance a .csv or .tsv file. The parameter -f describes which column will be extracted and can also be used to extract multiple columns.
# Look at some experimental metadata and extract the column we are interested in
less /nfs/teaching/551-0132-00L/2_Good_practices/metadata.tsv
# Extract the 4th column from left to right
cut -f 4 /nfs/teaching/551-0132-00L/2_Good_practices/metadata.tsv
# Extract multiple columns
cut -f 4,5 /nfs/teaching/551-0132-00L/2_Good_practices/metadata.tsv
The command paste allows you to put data from different files into columns of the same file.
# Put together two files into one
paste /nfs/teaching/551-0132-00L/2_Good_practices/sort_words.txt /nfs/teaching/551-0132-00L/2_Good_practices/sort_nums.txt
The command tr will replace a given character set with another character set, but to use it properly you need to know how to combine commands (below).
# For instance, this command requires you to type the input in
tr 'ABCDEFGHIJKLMNOPQRSTUVWXYZ' 'abcdefghijklmnopqrstuvwxyz'
# Then try typing AN UPPER CASE SENTENCE
# Remember to exit a program that is running use ctrl + c
# It can also be used to delete characters
tr -d 'a'
# Then try typing a sentence with the letter 'a' in it.
# Remember to exit a program that is running use ctrl + c
Advanced Exercise A1.2#
Advanced Exercise A1.2
Extract the second column of this file using cut.
# Extract the second column
cut -f 2 /nfs/teaching/551-0132-00L/2_Good_practices/sort_tab.txt
Use paste to combine the two files sort_words.txt and sort_nums.txt (in the directory
/nfs/teaching/551-0132-00L/2_Good_practices/) into a single two-column output.
# Use paste to combine files
paste /nfs/teaching/551-0132-00L/2_Good_practices/sort_words.txt /nfs/teaching/551-0132-00L/2_Good_practices/sort_nums.txt
Use tr so that when you enter the word banana it comes out as rococo.
# Use tr to convert one word into another
tr 'ban' 'roc'
# Then input banana and back comes rococo!
# Use ctr + c to kill the command
File permissions#
A very useful option for the command ls is -l, often given as alias ll. So if you use ll instead of ls it will not only list the file names within a directory but also give you information about file permissions, owner, file size, modification date etc.
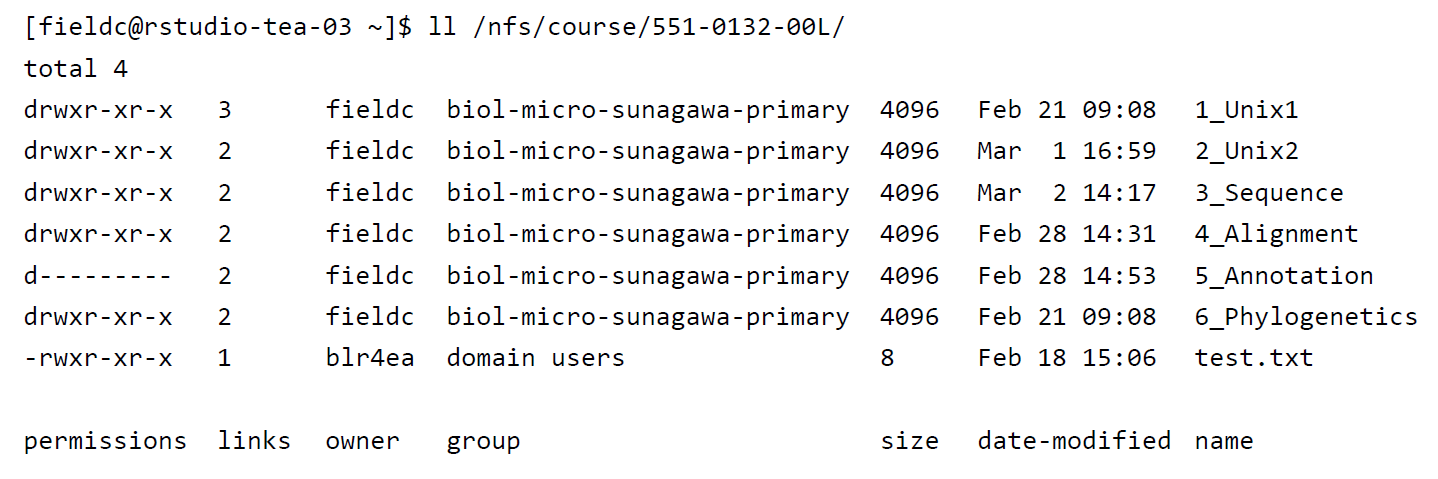
File permissions are useful to define who has access to read a file, write (make changes to the file) and execute (run a script) a file. Permissions are always split into 3 sections, the first section describing the permissions for the file owner, the second section describing permissions for a defined user group and the third section describing permissions for everyone else. Within each section, permissions are split into read r, write/edit w and execute/run x permissions.

# File permissions
drwxrwxrwx Any user (owner, user group, everyone) has permissions to read, write and execute a file
drwxr-xr-x The owner has permission to read, write, execute, while the user group and everyone only have read, execute permissions
drwxrwx--- The owner and user group have access to read, write and execute the file while others cannot access the file at all
Advanced Exercise A1.3#
Advanced Exercise A1.3
Write a simple script that will count the number of entries in a fasta file
Use a variable to allow you to declare the file when you run the script
# Use the text editor of your choice. Here we are going to use vim.
vim fastacount.sh
# Press "i" to enable the writing option and write a simple script to count fasta entries in a file.
grep -c "^>" $1
# You can exit your script by pressin "esc" and typing ":wq" (w for saving and q for quitting).
Make your script executable with the command “chmod +x”
# Make it executable
chmod +x fastacount.sh
Test it on some of the fasta files in the
/nfs/teaching/551-0132-00L/1_Unix/genomessubdirectories
# Run the script
./fastacount.sh /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna # 4302