Combining commands#
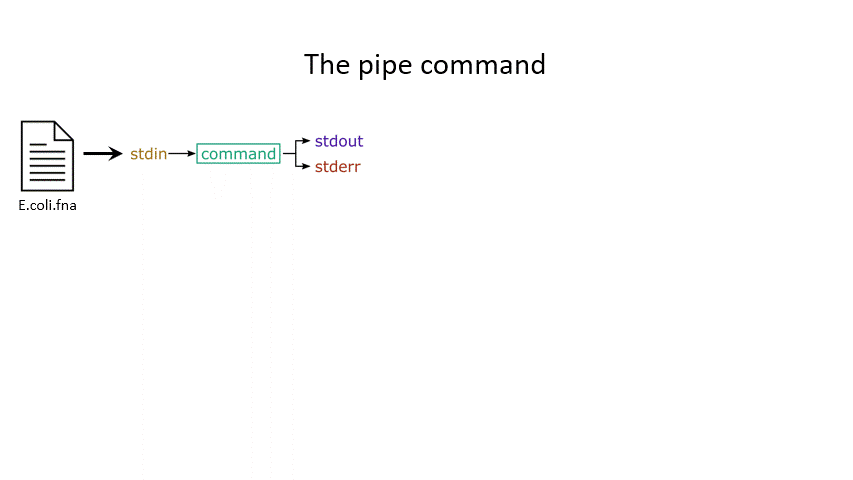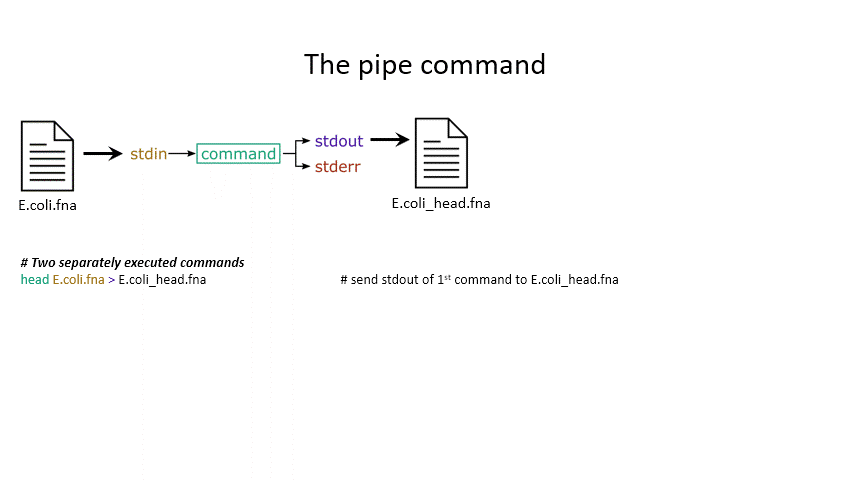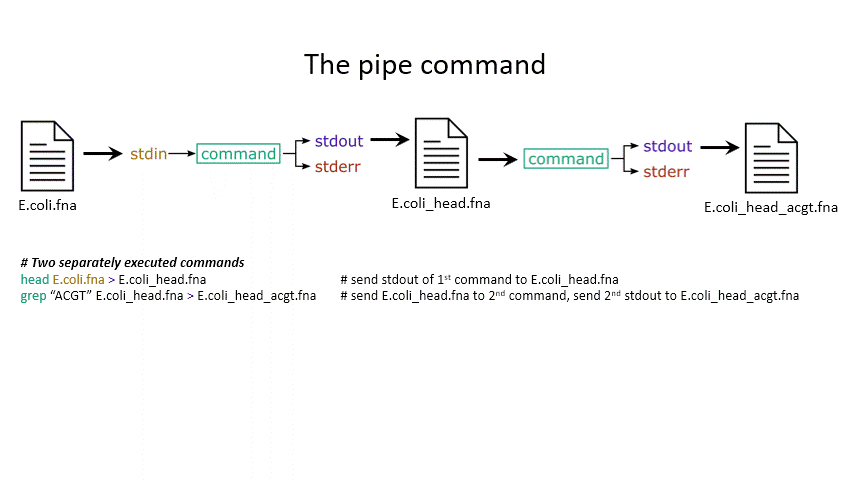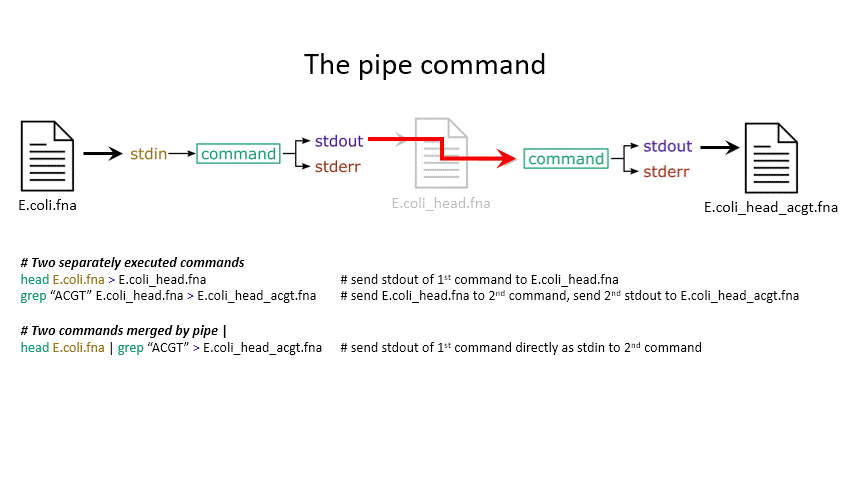
The power of the set of commands you learned about during the last practical comes when you use them together. Sometimes you want to take the output of one program and use it as the input for another – for instance, run grep on only the first 10 lines of a file from head. This is a procedure known as piping and requires you to put the | character in between commands (although this may not work with more complex programs).
# Copy and rename the file containing the E.coli open reading frames
cd
cp /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna E.coli_CDS.fna
# Piping
head E.coli.fna | grep "ACGT" # send the output of head to grep and search
grep -A 1 ">" E.coli_CDS.fna | grep -c "^ATG" # use grep to find the first line of sequence of each gene and send it to a second grep to see if the gene starts with ATG
Remember that “^” stands for the beginning of a line so the command shown above will only look for the pattern “ATG” at the beginning of a line. Also please remember, if you are ever unsure what a command does, try to get help from either manual/help pages or online to understand what each element of a command does.
Exercise 2.4#
Exercise 2.4
Rename the file GCF_000005845.2_ASM584v2_cds_from_genomic.fna (put in your home directory earlier) to E.coli_CDS.fna
# Go to your home directory
cd
pwd
# Copy the file to your home directory
cp /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna ~/E.coli_CDS.fna
Use grep to find all the fasta headers in this file, remember that a fasta header line starts with ‘>’.
# Find the fasta headers
grep '^>' E.coli_CDS.fna
Send the output of this search to a new file called cds_headers.txt.
# Send the output to a new file
grep '^>' E.coli_CDS.fna > cds_headers.txt
Use grep again to find only the headers with gene name information, which looks like, for instance [gene=lacZ], and save the results in another new file called named_cds.txt.
# Find named genes
grep '\[gene=' cds_headers.txt > named_cds.txt
Use wc to count how many lines are in the file you made.
# Count how many there are
wc -l named_cds.txt
Now repeat this exercise without making the intermediate files, instead using pipes.
# Repeat without intermediate files
grep '^>' E.coli_CDS.fna | grep '\[gene=' | wc -l