Gene prediction#
Gene prediction in prokaryotes#
In prokaryotes, the search for genes is relatively simple: As the coding density across a prokaryotic genome is relatively high, most of the genome sequence encodes genes thus there are small intergenic regions. Genes in prokaryotes are also relatively simple in structure: they begin with a start codon, end with a stop codon and contain a ribosomal binding site (RBS) motif. When looking for a gene, there are six possible reading frames, or six possible ways to convert a nucleotide sequences into an amino acid sequence:

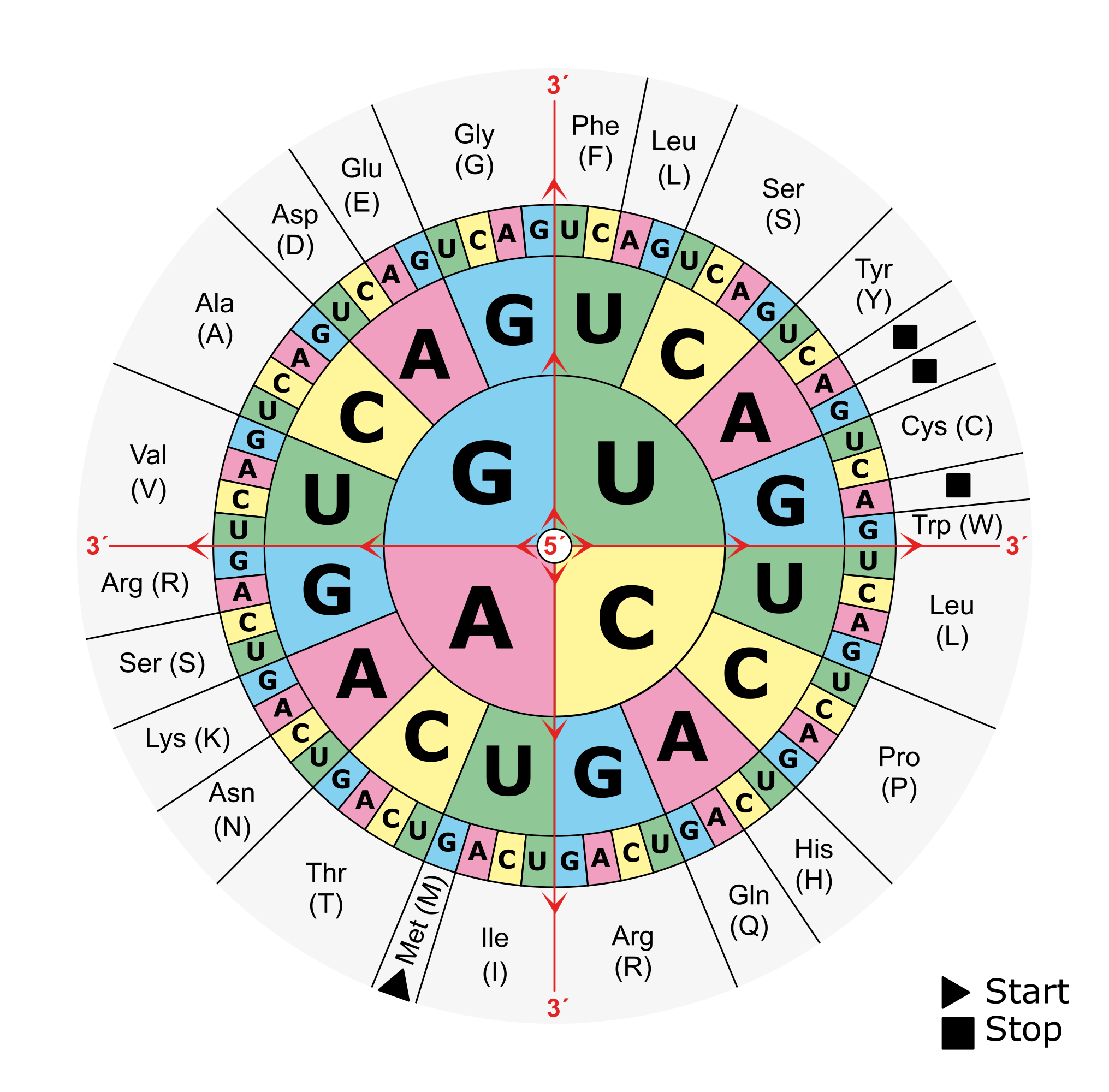
There are 3 frames on the top strand reading left to right, starting with the 1st, 2nd or 3rd base of the sequence in green, orange or magenta respectively. There are 3 additional frames on the complementary strand reading right to left, starting with the last, last but one or last but two base of the sequence in magenta, orange or green respectively. An open reading frame (ORF) is a region in one of these reading frames that begins at a start codon (M for Methionine) and then extends long enough that the region could be a gene before reaching a stop codon (* for any of the three possible stop codons). In this example a very small open reading frame exists, highlighted in grey. As a reminder, this is the amino acid codon table for prokaryotes, remembering that the base U in RNA corresponds to the base T in DNA:
Gene prediction algorithms look for these open reading frames and then use a model based on training data to determine which are likely genes, taking into account:
The start codon distribution
RBS motifs
Gene lengths
Overlap between genes (on the same and opposite strands)
Gene prediction in eukaryotes#
In eukaryotes, genes are more complicated features, notably with introns and exons that make the prokaryotic approach unworkable. The approach is instead to train a more sophisticated model on existing eukaryotic genes in a closely-related organism and use this model to predict genes in novel sequence. An example of software that does this is GlimmerHMM, available here. As we said at the start of the lesson Alignment within the section Comparative Sequence Analysis, it is beyond the scope of this course to look at this in more detail.
Prodigal#
The most popular prokaryotic gene predictor is a software package called prodigal, available here. We have also made it available on our module system (ml prodigal). Minimally, prodigal requires an input file in fasta format of at least 20kbp and will produce output directly to the command line. In our example command, instead of producing the output directly to the command line, we use the -o option to send the output to a file.
# Minimal example of running prodigal
prodigal -i my_genome.fasta -o my_genome.gbk
# Running prodigal with useful outputs
prodigal -i my_genome.fasta -o my_genome.gbk -d my_genes.fna -a my_genes.faa
In the second example we add the -d and -a options to output the nucleotide and amino acid sequences of the predicted genes to files. You can also modify the format of the main output file with -f and other options allow you to tailor the algorithm to your application, such as for a metagenome or just to train the algorithm parameters for use on another sequence.
Exercise 5.1#
Exercise 5.1
If given the amino acid sequence KVRMFTSELDIMLSVNGPADQIKYFCRHWT*, what is the number of amino acids in the encoded peptide (not including the stop codon)? Additonally, how many bases are contained in the open reading frame of the DNA sequence encoding the amino acids (including the stop codon)?
Your peptide contains 27 amino acids starting from the first methionine and ending with the stop codon *. Within your open reading frame there are 84 (28 x 3) bases.
Which specific codons might signal the termination of translation?
TAA, TAG and TGA are the three stop codons that initiate termination of translation.
Run prodigal on one of the genomes you have previously worked with, either in
/nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/or one you downloaded.
# Load the module
ml prodigal
# Run the program (change the filenames for your genome)
prodigal -i my_genome.fasta -o my_genome.gbk -d my_genes.fna -a my_genes.faa
Using command line tools, count how many genes were annotated (you can use any of the output formats for this but some are easier than others).
# Count the number of predicted genes
grep -c "^>" my_genes.fna
# Use seqtk on a multi-fasta file to receive the number of genes
ml seqtk
seqtk comp my_genes.fna | wc -l