Combining commands#
The power of this set of commands comes when you use them together, and when you can save your manipulated data into a file. To understand how to do this we have to think about the command line input and output data.
Input and output#
So far we have been using files as arguments for the commands we have practiced. The computer looks at the memory where the file is stored and then passes it through RAM to the processor, where it can perform whatever you have asked it to. We have seen output on the terminal, but it’s equally possible to store that output in memory, as a file. Similarly, if we want to use the output of one command as the input to a second command, we can bypass the step where we make an intermediate file.
The command line understands this in terms of data streams, which are communication channels you can direct to/from files or further commands:

stdin: the standard data input stream
stdout: the standard data output stream (defaults to appearing on the terminal)
stderr: the standard error stream (also defaults to the terminal)
Although you can usually give files as input to a program through an argument, you can also use stdin. Further, you can redirect the output of stdout and stderr to files of your choice.
# Copy and rename the file containing the E.coli genome
cd
cp /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.fna E.coli.fna
# Using the standard streams
head < E.coli.fna # send the file to head via stdin using '<'
head E.coli.fna > E.coli_head.fna # send stdout to a new file using '>'
head E.coli.fna 2> E.coli_err.fna # send stderr to a new file using '2>'
head E.coli.fna &> Ecoli_both.fna # send both stdout and stderr to the same file using '&>'
head < E.coli.fna > E.coli_head.fna 2> E.coli_err.fna # combine all three stdin, stdout, stderr
Chaining programs together#
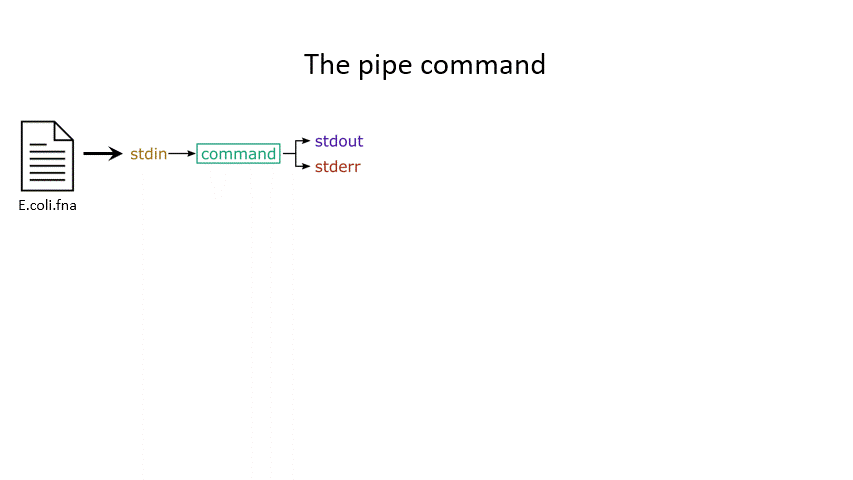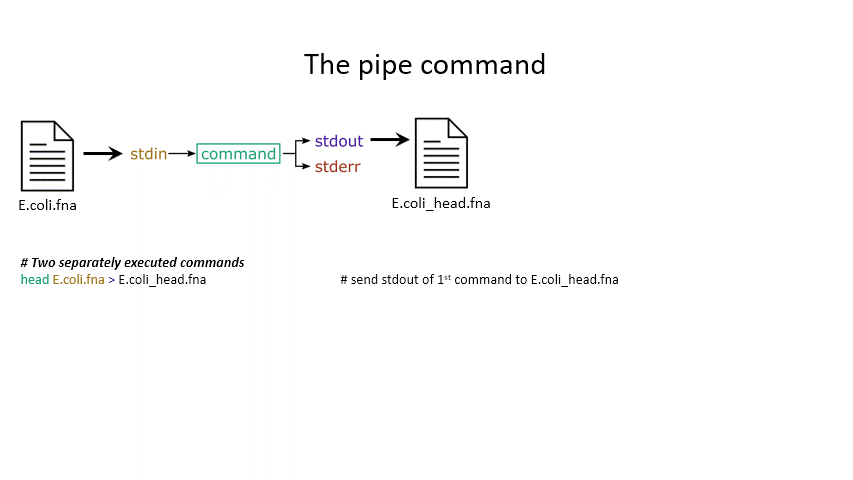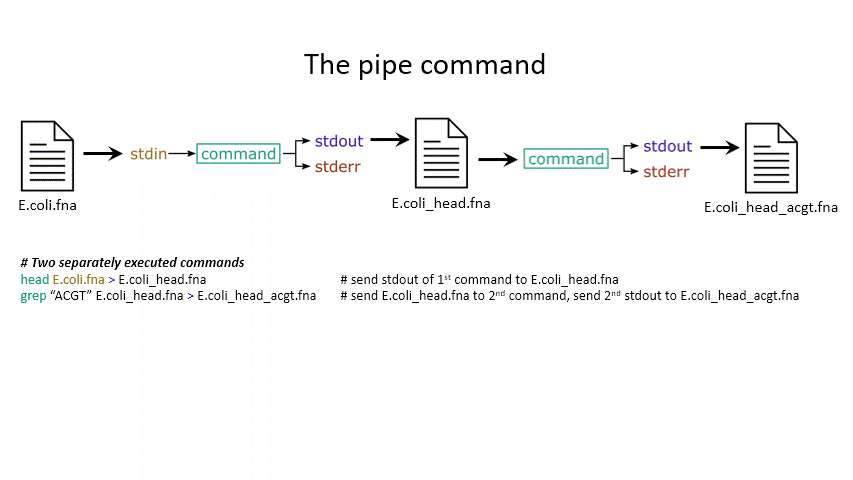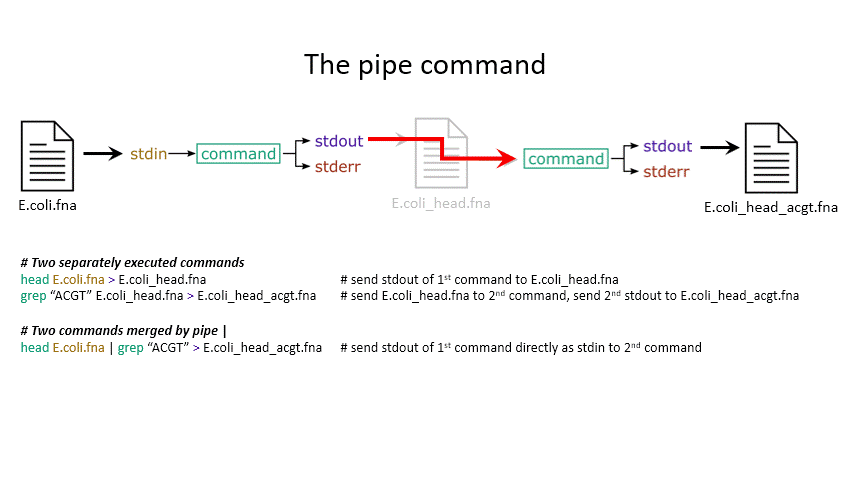
Sometimes you want to take the output of one program and use it as the input for another – for instance, run grep on only the first 10 lines of a file from head. This is a procedure known as piping and requires you to put the | character in between commands (although this may not work with more complex programs).
# Copy and rename the file containing the E.coli open reading frames
cd
cp /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna E.coli_CDS.fna
# Piping
head E.coli.fna | grep "ACGT" # send the output of head to grep and search
grep -A 1 ">" E.coli_CDS.fna | grep -c "^ATG" # use grep to find the first line of sequence of each gene and send it to a second grep to see if the gene starts with ATG
Exercise 0.8
Rename the file GCF_000005845.2_ASM584v2_cds_from_genomic.fna (put in your home directory earlier) to E.coli_CDS.fna
# Copy the file to your home directory
cp /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna ~/E.coli_CDS.fna
Use grep to find all the fasta headers in this file, remember that a fasta header line starts with ‘>’.
# Find the fasta headers
grep '^>' E.coli_CDS.fna
Send the output of this search to a new file called cds_headers.txt.
# Send the output to a new file
grep '^>' E.coli_CDS.fna > cds_headers.txt
Use grep again to find only the headers with gene name information, which looks like, for instance [gene=lacZ], and save the results in another new file called named_cds.txt.
# Find named genes
grep '\[gene=' cds_headers.txt > named_cds.txt
Use wc to count how many lines are in the file you made.
# Count how many there are
wc -l named_cds.txt
Now repeat this exercise without making the intermediate files, instead using pipes.
# Repeat without intermediate files
grep '^>' E.coli_CDS.fna | grep '\[gene=' | wc -l
As an additional challenge:
Using the commands we have used, find the start codon of each gene in E. coli and then count up the frequency of the different start codons.
# Count the frequency of start codons in the *E.coli* genome
grep -A 1 '^>' E.coli_CDS.fna | grep -Eo '^[ACGT]{3}' | sort | uniq -c | sort -nr -k 1
# The first part finds all headers plus the first line of sequence
# The second part is a regular expression to find the first three nucleotides in the sequence lines
# Then we have to sort them so that we can count them with uniq
# The final part is a bonus that sorts by descending frequency
# And as so often in bioinformatics, there are several ways of getting a task done.
# Consider the following alternative:
grep -A 1 ">" E.coli_CDS.fna | grep -v '>' | grep -o "^\w\w\w" | sort | uniq -c | sort -k1nr
Writing and running a script#
If you construct a series of commands that you want to perform repeatedly, you can write them into a script and then run this script instead of each command individually. This makes it less likely that you make an error in one of the individual commands, and also keeps a record of the computation you performed so that your work is reproducible.
You can write the script using a text editor on your computer and then uploading it with scp (for examble notepad). If you want to write a script directly in the terminal there are text editors available such as vim and emacs - you should be able to find tutorials for both online (for example openvim or GNU Emacs).
By convention, a script should be named ending in .sh and is run as follows:
# Run a script in the same directory
./myscript.sh
# Run a script in another directory
./mydir/myscript.sh
The command line interface, or shell, that we use is called bash and it allows you to execute the same script with changing arguments, encoded as variables $1, $2, etc.
For instance we could have a simple script:
# myscript.sh
echo "Hello, my name is $1"
# Running my simple script
./myscript.sh Chris
"Hello, my name is Chris"
This means you could write a script that performs some operations on a file, and then replace the file path in your code with $1 to allow you to declare the file when you execute the script. Just remember that if your script changes working directory, the relative path to your file may be incorrect, so sometimes it is best to use the absolute path.
File permissions#
A very useful option for the command ls is -l, often given as alias ll. So if you use ll instead of ls it will not only list the file names within a directory but also give you information about file permissions, owner, file size, modification date etc.
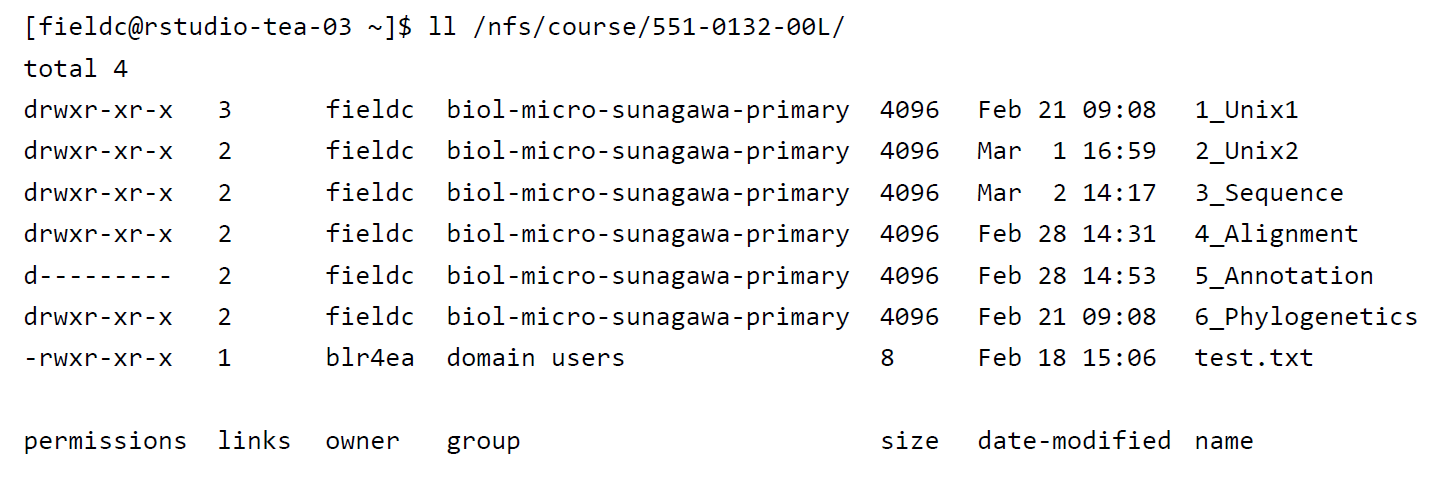
File permissions are useful to define who has access to read a file, write (make changes to the file) and execute (run a script) a file. Permissions are always split into 3 sections, the first section describing the permissions for the file owner, the second section describing permissions for a defined user group and the third section describing permissions for everyone else. Within each section, permissions are split into read r, write/edit w and execute/run x permissions.

# File permissions
drwxrwxrwx Any user (owner, user group, everyone) has permissions to read, write and execute a file
drwxr-xr-x The owner has permission to read, write, execute, while the user group and everyone only have read, execute permissions
drwxrwx--- The owner and user group have access to read, write and execute the file while others cannot access the file at all
Exercise 0.9
Write a simple script that will count the number of entries in a fasta file
Use a variable to allow you to declare the file when you run the script
# Use the text editor of your choice. Here we are going to use vim.
vim fastacount.sh
# Press "i" to enable the writing option and write a simple script to count fasta entries in a file.
grep -c "^>" $1
# You can exit your script by pressin "esc" and typing ":wq" (w for saving and q for quitting).
Make your script executable with the command “chmod +x”
# Make it executable
chmod +x fastacount.sh
Test it on some of the fasta files in the
/nfs/teaching/551-0132-00L/1_Unix/genomessubdirectories
# Run the script
./fastacount.sh /nfs/teaching/551-0132-00L/1_Unix/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna # 4302