Computing at the Institute of Microbiology#
Learning objectives#
This workshop introduces FAIR principles and covers effective ways of managing your data and code:
What FAIR principles are
How to structure your work on the computer
Which files should be backed up, which should not be
Using Git, Github, Gitlab
Preparing your work for publication
Archiving your data and code for the future
Introduction#
The aim of this workshop is to encourage good practice when undertaking computer-based work. Here we will focus on organisation and documentation, but if you are interested in, for instance, how to be a consdierate user of computing servers, please see the previous workshop.
If you are interested in a more detailed course on the topic of data stewardship, I recommend this in-depth course run by the ETH library.
FAIR principles#
FAIR data is defined as Findable, Accessible, Interoperable and Reusable, which are principles designed and endorsed by scientists in a 2016 paper.
By SangyaPundir - Own work, CC BY-SA 4.0, https://commons.wikimedia.org/w/index.php?curid=53414062#
In brief:
Findable data is well-described, well-referenced, assigned a unique and persistant identifier, and registered in a searchable resource.
Accessible data is retrievable by a standard protocol that is open and free, and allows for authentication when necessary.
Interoperable data is machine-readable, and uses standardised language and terms (such as defined by a FAIR ontology).
Reusable data includes sufficient metadata and rights to allow use by another, and uses open, long-term viable file formats.
Some of this sounds quite daunting, and indeed it is a lot of work to make the data from a large project conform to all of these principles. It is easy, however, to make simple decisions that will make your data come closer to these standards. For instance:
Naming your files appropriately and using a clear file structure for storage
Choosing to use file types that are open (i.e.: not Microsoft Excel)
Documenting all the commands, scripts and code that went into your data analysis so that your entire pipeline can be reproduced
Submitting your data to appropriate online resources
What we hope to cover here is what you can do on a practical level to aim towards these principles.
Data Generation Exercise
Make a list of all the types of files you create during your work
Think about each file type. Is it FAIR?
How can you make the unsuitable data conform to FAIR standards?
Structuring your work#
In the Sunagawa lab, we have established a project-based directory structure in our computer-based work, elements of which mirror the way we organise electronic lab notebook entries. This is useful because it ensures that we efficiently use the different storage space available to us - backed-up vs. not - and also provides a framework for later publishing and archiving the work.
It follows this structure, with directories in [brackets] as optional but sometimes useful:
.
├── code
├── data
│ ├── raw
│ ├── [output1]
│ ├── [output2]
│ └── ...
├── [lib]
├── [logs]
├── scratch
│ ├── [output1]
│ ├── [output2]
│ └── ...
├── LICENSE
├── README.md
└── [PROGRESS]
code is a directory for your scripts
data is for storing initial raw data, and processed data that you want to keep
lib is optional, for storing reference data
logs is optional, for keeping logs of software that you have run
scratch is for experimenting in, for putting processed data that you aren’t sure yet if you want to keep
LICENSE is a file we add by default so that the terms of using the data are clear
README.md is a markdown file describing the project
PROGRESS is an optional file for tracking project milestones and keeping notes
Any of the primary folders can contain subfolders as needed, with those in data and scratch above as examples. If you perform the same computation on multiple initial samples, it’s a matter of taste whether you organise your subfolders by sample, then processing stage, or by processing stage, then sample.
If you create this structure in your Work Folders or on your group’s gram drive, everything will be automatically backed up. In this case, it would make sense to keep an eye on how much space you are taking up on scratch so as not to waste it. For other forms of storage and later stages of the project, you should carefully decide what data belongs in data rather than scratch, and likely only keep backups of code, data and logs, as well as the text files. In fact, whatever ends up in data may best be submitted to online resources for access and long-term storage (and is likely to be what ends up in your publication).
Tips for working on a server#
One of the nice features of working on a Linux server is that it saves a record of the commands you have run, up to a certain limit. They are saved in a hidden file in your home directory, .bash_history and accessible on the command line by the up and down arrow keys. You can also search the history by pressing Ctrl + r, but it takes a bit of getting used to.
You can increase how many lines are kept in this history by modifying another hidden file in your home directory, .bashrc, in which all commands are run whenever you start a new session.
PROMPT_COMMAND='history -a' # adds lines to the history file immediately rather than when a session ends
HISTTIMEFORMAT="%F %T " # adds timestamps to the history file
HISTSIZE=10000 # increases the accessible history size to 10000 lines
HISTFILESIZE=20000 # increases the history file size to 20000 lines
Another good practice is to keep logs of the commands you run. If you put jobs into a server queue, then make sure to declare where you want the log files to be saved. If you are just running something casually, you can direct the output that normally appears to log files of your choosing:
# Direct standard out
./myscript.sh > out.txt
# Direct standard error
./myscript.sh 2> err.txt
# Direct both to separate files
./myscript.sh > out.txt 2> err.txt
# Direct both to the same file (can be messy)
./myscript.sh &> outerr.txt
Git#
Git is a version control system that tracks changes in a specified set of computer files. You may have heard of either Github or Gitlab, which are two platforms that support the protocol. The Gitlab linked to here is run by ETHZ, whereas Github is a commercial enterprise, but offers a good amount of free use and other advantages. Both the Sunagawa and Vorholt groups maintain organisational accounts, and one also exists for the institute itself.
.gitignore#
As part of our project system, we create a Github repository. We don’t want all files to be tracked: scratch especially not, but likely also data and lib since they will contain large files. To prevent such files being included in our repository, we add the paths to a special hidden file called .gitignore (the initial . designates the file as hidden). For instance:
cat .gitignore
/data/
/scratch/
/lib/
When you create a repository (see below), you can automatically generate a file based on templates from github, or use a tool such as this one.
Creating a repository#
From this point, we will work on Github, so you will need to sign up for an account. The most straight-forward way to create a repository is on this page. It requires:
A name - we recommend the same name as the project directory on your file system
A description - something that will allow you to identify the project
Public or Private - initially your repository should be private until you are ready to publish your work
Optionally, you can tick the box Add a README file but best not to if you have already created your own
You can also choose a pre-configured .gitignore file for the programming language you’re using, but you still ought to modify it as above
You can also choose a licence - we recommend the GNU General Public License v3.0
Once you have created the repository, it will be online at github/<username>/<reponame>.
Now, to connect it with the project directory on your linux system, do the following (don’t worry about the meaning of the commands for now):
# Navigate to your project folder
# Initialize the local repository
git init -b main
# Make sure that you have any files created in the initial remote repo
git pull https://github.com/<username>/<reponame> main
# Now add your files to the local repo
git add . # or
git add --all
# Commit the files
git commit -m 'First commit'
# Connect the local with the github repo - fill in your username and reponame
git remote add origin https://github.com/<username>/<reponame>
# Verify the URL
git remote -v
# Push your first commit
git push origin main
If you are working on Windows, you can use the Github desktop client.
Using git#
By Daniel Kinzler - Own work, CC BY 3.0, https://commons.wikimedia.org/w/index.php?curid=25223536#
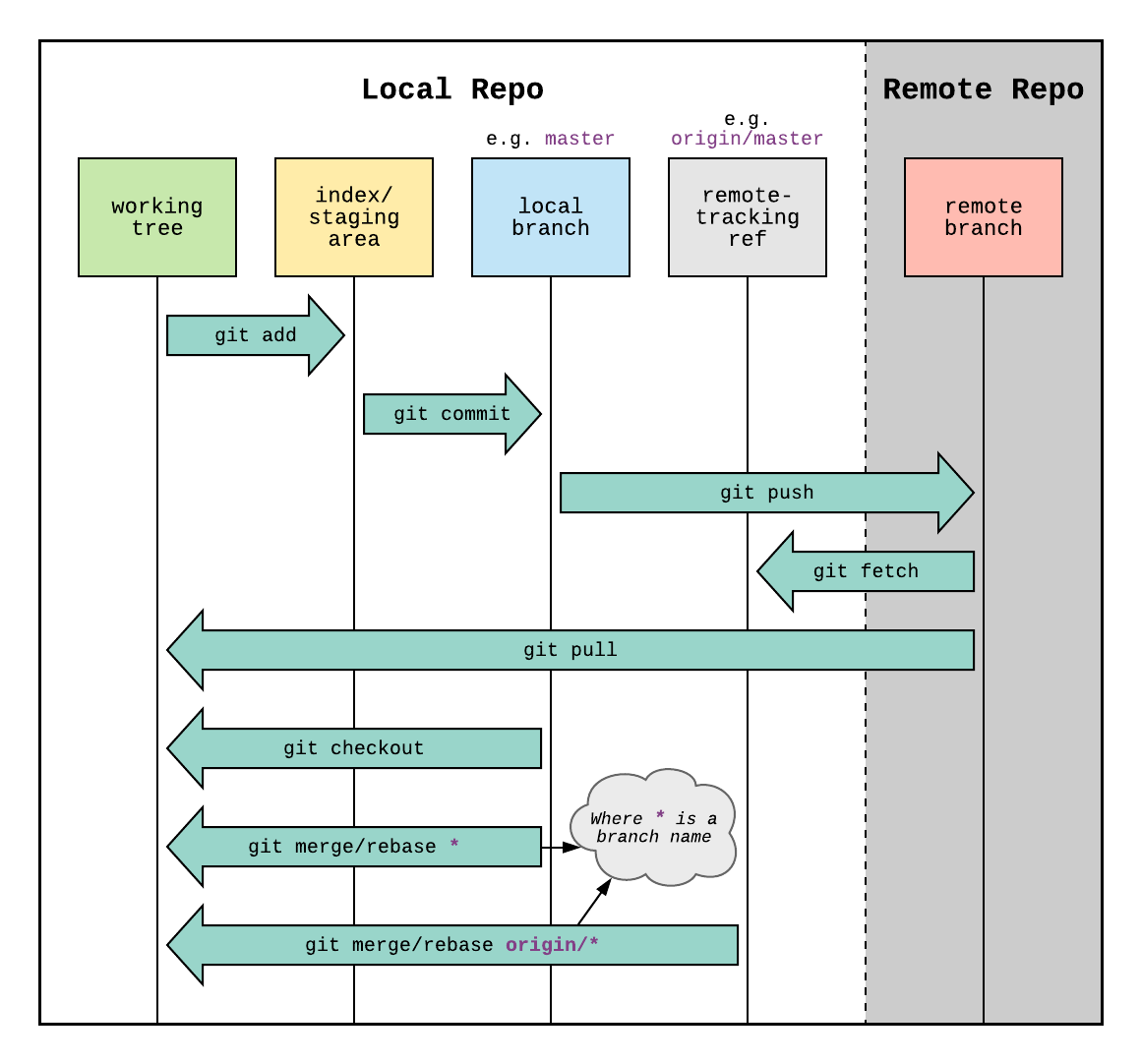
Generally, once you have set up your repository, you will over time make changes to the files there as you work. At some point you will want to make a commit - recording those changes and synchronising the remote repository with your local version. The first three arrows on this diagram describe this process:
git add- choosing files to be tracked for this commitgit commit- recording the file changes into a formal commit (often with an associated message-m "my commit"git push- pushing those changes to the remote repository
The reverse, synchronising a local repository with the remote version is performed with git pull.
Another useful command is git clone, allowing you to create a copy of the remote version fresh on your local system (no local repo setup required).
Really, this is all you need to know to ensure you keep your work backed up. You can even work between multiple machines this way, so long as before you start to make any changes to your local repo you make sure to git pull and synchronise with the latest remote version. If you start to work with others on the same repository at the same time, you will need to read about merging, and better still understand branches, but those are topics for another workshop.
Publishing your work#
When it comes to publishing your work, different journals and funding bodies have different requirements. What we recommend is to go well beyond these requirements and to provide everything that is needed to make your data FAIR - another scientist should be able to read your paper, then download and run your data through the same code you did, and get the same results (or at least draw the same conclusions if there are stochastic elements in your work). It might take some effort but try to follow these steps:
1. Reorganise your work#
If your project wasn’t too complicated or you’ve been very good, you won’t have to worry about this. Often though, you have done analyses that don’t make it into your publication or created intermediate filesthat aren’t worth keeping. To reorganise your work:
Start a fresh project directory
Copy over the code, initial data and any intermediate files that you don’t want to recreate
Run the code to check it works with the data in the new project
Take the opportunity to tidy the code:
Only keep the parts that make output you want to publish
Separate the code into units that correspond with your publication
Comment on the code where you have previously failed to
Ensure the code for your figures is also clear and available
When the tidy-up is done, you should write appropriate metadata files to describe what your repository contains - what is each file, what format is it and how was it created. You can then create the associated github repo ready for publication.
2. Submit data to online resources#
We recommend submitting sequence and related data to the European Nucleotide Archive (ENA). Documentation for this is hosted here, but we are happy to help you with this as we have a great deal of experience. The data may include:
Raw sequencing reads
Genome and metagenome assemblies
Annotated genomes and metagenomes
Metagenome bins and Metagenomically Assembled Genomes (MAGs)
Of these, annotations are the most difficult to make compatible with the requirements. In particular, analyses such as performed by antiSMASH are not compatible. These you may want to publish separately (see below).
Everything you have submitted should have an associated reference that you can include in your publication.
3. Submit everything else to Zenodo#
Zenodo is a general repository for scientific research, data, software and the like, operated by CERN. There you can create a snapshot of your github repository to associate with your publication, which will grant it a DOI for permanent citation. You can also create an independent repository for data files that have no appropriate online resource to host them.
A quick start guide to Zenodo is available here.
4. Describe your methods in detail#
In the Methods section of your publication, describe in understandable terms what you did to process your data and reference your submitted data. In particular mention software versions in as much detail as possible. For Python and R there are some useful commands to let you describe the environment in which you ran your scripts, which can be placed in metadata files.
# For python, this will list all packages and versions
pip list
# In R, this will list all loaded packages and versions
sessionInfo()
Publication Data Exercise
Think about publications you have contributed to. What data did you make available?
How difficult do you think it would be to reanalyse your data to produce the same results/conclusions?
Is your current work organised? Do you want to spend the time organising it now rather than later?
Archiving your work#
When you’re approaching the end of your time at the institute, it’s helpful to archive your work in such a way that your group can easily access it. This basically follows the same principles as for publishing your work, but we have some different systems in place. Most of the task is organisation, see above, but then consider:
1. Check your raw data is stored#
Your original raw sequencing data should be submitted to Harvest.
2. Check your processed data is stored#
If you have processed data that hasn’t been submitted to an online resource (note that it does not need to be published for this) then make sure it’s in an appropriate place on your group’s gram drive with metadata describing each file and perhaps referring to how it was created.
3. Archive your projects#
After you have reorganised your projects, you can archive them. Simply compress each project directory and then follow the instructions to store them in the institute’s Long Term Storage (LTS).
If you are working on Windows, you can just zip them. If you are working on Linux, we recommend tarring and gzipping them.
Once again, ensure that each project is well documented with appropriate metadata. Then your final metadata file should describe each project, its contents and context.