Technical Workshop: Downstream 16S Analysis¶
Information¶
When: Wednesday 17th May, 2023 1pm-3pm
Where: G443
What: Ways to look at 16S data from a count table onwards
How: Bring a computer that can connect to the wifi
Learning Objectives¶
You will be able to:
import the ASV and OTU tables into R for analysis
normalize your data and understand when to use different methods
calculate measures of alpha and beta diversity, and differential abundance
visualise your data
Importing data¶
For this workshop, we will use some example data from the PHRT project, which can be found in the path /nfs/teaching/workshops/downstream_16s_analysis/.
The output of our standard 16S pipeline is as follows:
├── 0raw
├── 1cutadapt
├── 2filterAndTrim
├── 3learnerrors
├── 4sampleInference
├── 5mergeReads
├── 6bimeraRemoval
├── 7taxonomy
├── 8uparsetax
├── scratch.asvs.fasta
├── scratch.asvs.tsv
├── scratch.benchmark
├── scratch.command
├── scratch.done
├── scratch.insert.counts
├── scratch.log
├── scratch.otus.fasta
├── scratch.otus.tsv
└── scratch.otus.uparse
Two of these are count tables with metadata: scratch.asvs.tsv determined by dada2 and scratch.otus.tsv determined by uparse.
Exercise 1
Try to import these two files into R using
read.table.
asvs <- read.table("scratch.asvs.tsv", sep="\t", header=T)
otus <- read.table("scratch.otus.tsv", sep="\t", header=T)
We’ll now work with the ASV table, but everything we look at can be applied to both tables. If you inspect the columns of these table, you will see that the first few are not counts, but information about the ASVs. We need to split the count data from this metadata, creating two new variables to contain the data. The first column of the table would also be more useful if split in two, name and size, and if size were just a number. Finally, it would be good to have the ASV names as row names to make indexing easier later.
Exercise 2
Put the metadata columns into one data frame and the count data into another:
Split the first column with
strsplitConvert the new “size=xxx” column into numbers with
substringandas.numeric
Name the rows appropriately.
split_cols <- as.data.frame(do.call(rbind, strsplit(asvs$asv, ";")))
split_cols[,2] <- as.numeric(substring(split_cols[,2], 6))
colnames(split_cols) <- c("asv", "size")
asvs_meta <- cbind(split_cols, asvs[,2:5])
rownames(asvs_meta) <- asvs_meta[,1]
asvs_count <- asvs[,6:ncol(asvs)]
rownames(asvs_count) <- asvs_meta[,1]
Normalisation¶
Now our data is ready to work with. Firstly, let’s have a look at the sequencing depths of our samples.
colSums(asvs_count)
You can see that they are not equal. It would be unfair to make comparisons between samples without correcting for this with a normalisation procedure. One way to do this is with rarefaction, in which samples are randomly resampled down to a given depth - typically that of the shallowest sample, but some samples may have to be discounted for this to still be a decently high number. Alternatively, there are multiple methods that try to account for the statistical distribution of the data and model accordingly, for instance DESeq2.
Rarefaction¶
To rarefy our data, we will use the vegan package, however it assumes that our data is arranged with samples in rows and ASVs/OTUs in columns, so we also need to transpose the data. For example:
# load the library
library("vegan")
# rarefy data
asvs_rare = t(rrarefy(t(asvs_count), sample_size))
The choice of sample_size is a difficult one. You should identify samples that you can definitely remove or use in other analysis, such as controls, or ones that are significantly shallower and would involve throwing away too much data to include. There are no fixed rules for choosing how low to rarefy.
Exercise 3
Filter your data table (and accompanying metadata) to remove low depth and control samples
Rarefy your data to a round number, as deep as possible with the data you kept
Take a look at one sample in your rarefied data - note how many ASVs appear with counts above zero
Rarefy your data again and repeat this exercise, is there any difference?
If you repeat the rarefaction you will likely see a different result because this process is random. This means that for low abundance ASVs, they may sometimes appear present and sometimes absent, which could affect your analysis. If you are unsure how robust any conclusion is as a result of rarefaction, you can always perform the calculations many times and summarise the final results across all rarefactions.
To get an idea of whether your rarefied data is representative of the samples, we can also plot a rarefaction curve:
# plot curves
rarecurve(t(asvs_count), step=1000, label=F)
# add line to show rarefaction
abline(v=sample_size, col="red")
You can see the depth to which you have rarefied by the red line. Ideally the line is intersecting the sample curves when they are relatively horizontal.
DESeq2¶
Developed for normalising RNASeq samples, DESeq uses a statistical model to estimate the actual abundance of each sequence so that samples can be compared against each other even though they may be of different depths. We won’t go into the details here, but the model assumes that many sequences do not change abundance across most samples, which may not be true for your data. Additionally, if a sequence doesn’t appear in a particular sample, then it provides an estimate of how abundant that sequence was instead of assuming zero. This makes certain types of downstream analysis useless. However, when you are working with a synthetic community, where it is reasonable to assume that all species ought to appear in your results, even at very low abundance, then this is not so bad.
One key point to understand about 16S sequencing data is the mean-variance relationship. A species with low abundance, around the detection threshold, has a high count variance compared to its mean - that is, if you were to sequence the same sample multiple times you would see very low values but the variance between those counts would be high. Also, as you look at species with higher and higher abundance, the variance scales with the mean, increasing accordingly. Most model-based methods try to correct for this relationship in some way.
If you are interested in using DESeq2 then there is a comprehensive vignette available.
Other methods¶
Other model-based methods include:
limma-voom
metagenomeSeq
edgeR
suggestions please
Diversity¶
In ecology, the diversity of a community, or for us a sample, refers to various different measures. They can be classified as measures of either alpha diversity, which is calculated on a single sample, or beta diversity, which is calculated by comparing multiple samples.
Alpha measures¶
Two basic measures of a sample’s species diversity are:
Richness - the number of species present in a sample
Evenness - how equal the abundances of different species in a sample are
For 16S data, we define species by our ASVs, so the richness is an easy calculation:
# calculate species numbers
spec_rich <- specnumber(t(asvs_rare))
However there are measures that attempt to calculate the true richness of a sample, such as the Chao1 index:
# calculate various richness measures
multi_rich <- estimateR(t(asvs_rare))
For evenness, there are many measures to choose from. We would routinely consider the Shannon, Simpson and Pielou diversity indices.
The Shannon index is calculated as follows, where i stands for each species in turn of R species, \(p_{i}\) stands for the relative abundance of species i in the sample, and ln is the natural logarithm.
# calculate shannon diversity
shannon_even <- diversity(t(asvs_rare), index="shannon")
The Pielou index is a modification of this to represent the ratio between the observed Shannon index and the Shannon index if all species were equally abundant.
# calculate pielou diversity
pielou_even <- shannon_even/log(spec_rich)
The Simpson index and inverse are calculated based on this equation, where the index is \(1-D\) and the inverse is \(1/D\):
# calculate simpson index
simpson_even <- diversity(t(asvs_rare), index="simpson")
# calculate inverse simpson index
invsimpson_even <- diversity(t(asvs_rare), index="invsimpson")
Exercise 4
Calculate a richness and evenness statistic of your choice for the rarefied ASV data.
Plot these statistics in a visually appealing way - you may want to sort them, use colours to indicate metadata elements and so on.
Beta measures¶
To compare multiple normalised samples, we require a distance metric that defines how we weight differences in abundance between species and their presence/absence. The main function for this provided by the vegan package is vegdist, which provides a large number of options, see here.
Some metrics will not work with some kinds of normalisation - for instance, the popular bray-curtis dissimilarity will not work with data that has been log-normalised (for instance by DESeq2) as it cannot handle negative values.
There are different reasons for choosing different metrics. For instance, Euclidean distance is very simple to understand and compute, but is heavily biased by highly abundant species if you use rarefaction. It is very reasonable for data normalised by DESeq2 however, as it attempts to make the mean-variance relationship equal at all abundances.
Let’s look at the Bray-Curtis dissimilarity as an example:
# calculate bray-curtis dissimilarity matrix
bc_dist <- vegdist(t(asvs_rare), method="bray")
Visualisation¶
Here we will focus on ordination techniques - methods to transform multidimensional data such that the similarity and/or dissimilarity between objects is emphasised. Multidimensional scaling is the term used to cover a variety of ordination techniques that reduce the dimensionality of the data, especially so that it is useful to visualise in 2D.
PCA¶
Principle Component Analysis is a linear transformation that transforms the data to a new coordinate system where the first dimension captures the most variance, the second the second most and so on.
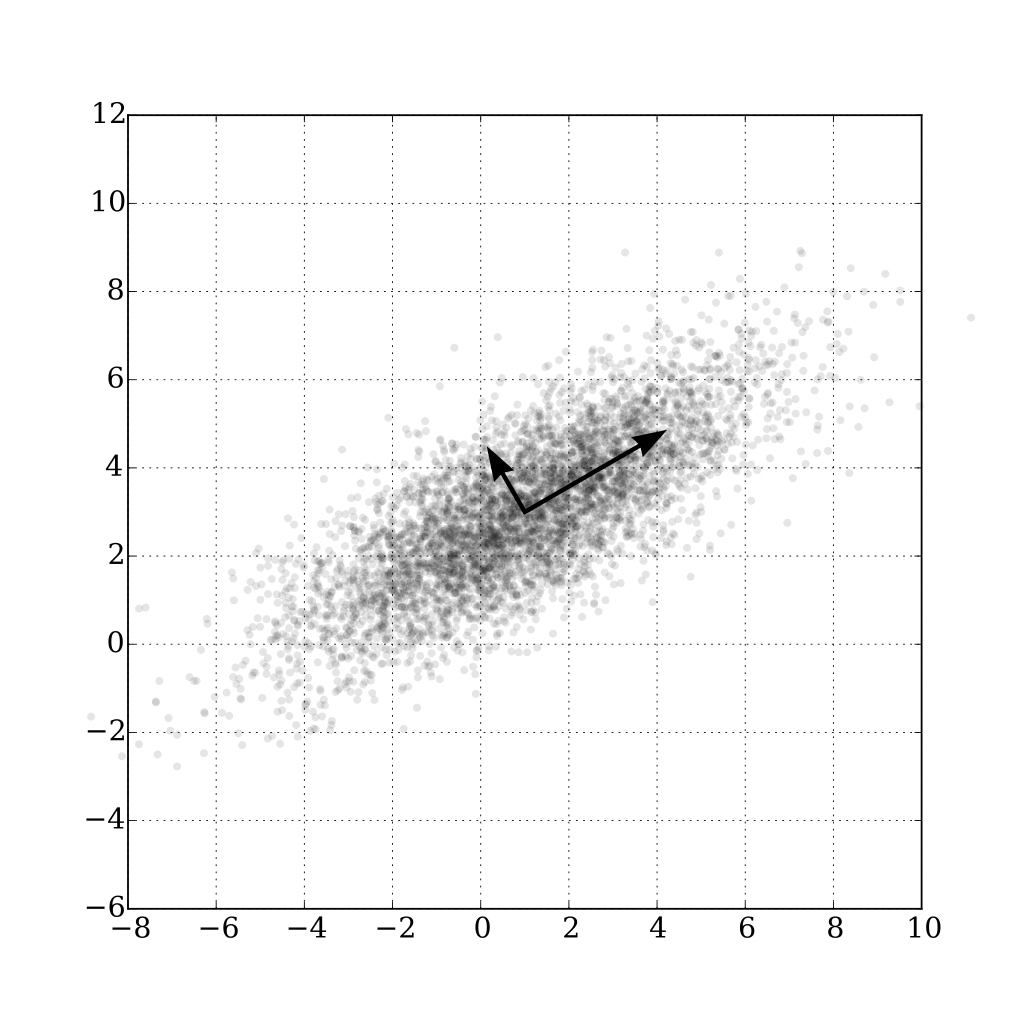
This is best used on the normalised count table, or a simple transformation of it such as Hellinger. Similar samples will appear close to each other, and dissimilar ones far from each other. This can allow you to visually cluster samples, or consider whether the first two principle components (the x and y axes) correspond with your metadata.
# calculate and plot the PCA
pca <- prcomp(asvs_rare)
biplot(pca)
# Hellinger transform
asvs_rare_hell <- t(decostand(t(asvs_rare), method="hellinger"))
pca_hell <- prcomp(asvs_rare_hell)
biplot(pca_hell)
PCoA¶
Principle Coordinate Analysis is effectively the use of principle component analysis on a distance matrix. It will also show similar samples close together and dissimilar ones far apart, and the axes may correspond to your metadata.
# calculate and plot PCoA
library(ape)
pcoa <- pcoa(bc_dist)
biplot(pcoa)
NMDS¶
Non-metric Multidimensional Scaling uses a non-parametric (distribution free) method to rescale the data. Unlike PCoA, it doesn’t ensure that the ordination distances between samples correlate with the original distances, only that the rank order is maintained. This can provide a better visual understanding, but distances are more distorted.
# calculate and plot NMDS; k is the number of reduced dimensions and you can experiment with changing it
nmds <- monoMDS(bc_dist, k=3)
plot(nmds, choices=c(1,2), type="t")
Others¶
There are an endless number of ordination techniques - each is a trade off between visual appeal and maintaining true distances between objects. For instance, the popular transform t-SNE can create beautiful visual clusters, but is highly misleading. Be wary of basing any conclusions on such techniques.
Exercise 5
Plot the PCA of the rarefied ASV data with and without Hellinger transformation.
Calculate the Bray-Curtis dissimilarity matrix for the same data.
Plot the PCoA and NMDS of this distance matrix.
Make a visually appealing plot of one of these analyses.
Differential abundance¶
So far we have looked at the data on a sample basis, but we can also consider differences between samples on an ASV basis. This is particularly tricky for 16S count data because it is compositional, which means, for instance, that if one ASV increases in relative abundance, others must necessarily decrease. Statistically speaking, this is a difficult problem to solve, which is why we often resort to non-parametric statistical tests - they are not as powerful but do not assume an underlying distribution.
A useful test is the Wilcoxon rank-sum test. Let’s not look at the maths this time and go straight to the code:
# we need to identify two groups of samples for the test
sushi_samples <- grepl('SUSHI', colnames(asvs_rare))
novo_samples <- grepl('NOVOGENE', colnames(asvs_rare))
# test for differential abundance in the first ASV
wilcox_result <- wilcox.test(asvs_rare[1, sushi_samples], asvs_rare[1, novo_samples])
wilcox_result$p.value
Exercise 6
Write a loop to obtain the p-value for each ASV
Perform p-value correction with
p.adjustAre any ASVs significantly different between the two sample sets?
wilcox_results <- rep(NA, nrow(asvs_rare))
for(i in 1:nrow(asvs_rare)){
wilcox_results[i] <- wilcox.test(asvs_rare[i, sushi_samples], asvs_rare[i, novo_samples])
}
wilcox_