Introduction to Unix 2¶
General information¶
Topic overview¶
In this lesson you will continue learning to use Unix-based systems. You will learn how to look at files and search them, how to control the flow of input and output to programs and responsible use of HPC clusters.
Learning objectives¶
Learning Objective 1 - You are able to observe, extract and manipulate data from files
You use commands such as less, cut and grep
You read command manuals and search for solutions online
You implement regular expressions
Learning Objective 2 - You can automate computational tasks and construct workflows to obtain reproducible results
You redirect the output and error streams to files
You pipe the results of one command to another
You write simple scripts and execute them
Learning Objective 3 - You are able to use High Performance Computing clusters (HPCs) to solve bioinformatic problems
You use a software module system
You manage software dependencies and resource requirements as part of a job submission script
You execute a job submission script on an HPC and inspect its output
Searching¶
Searching for a file¶
When you are trying to find a file in your system, the command find offers a number of options to help you. The first argument is where to start looking (it looks recursively inside all directories from there), and then an option must be given to specify the search criteria.
# Finding files ("." stands for the current directory you are in)
find . -name "*.txt" -type f # searches for files ending in .txt. The type option defines the type of the file.
find . -mtime -2 # searches for files modified in the last two days
find . -mtime +365 # searches for files modified at least one year ago
find . -size +1G # searches for files at least 1GB or larger
find . -maxdepth 1 # searches only on one level, thus only here, i.e.: doesn't look inside directories
Exercise 2.1
Use cp to copy all files from the directory
/nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2into a new directory in your home directory
# Make a directory for the new files
cd ~
mkdir ecoli
# Copy all the files
cp /nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/* ~/ecoli/
Navigate to the
/nfs/teaching/551-0132-00L/1_Unix1/genomes directory
# Navigation
cd /nfs/teaching/551-0132-00L/1_Unix1/genomes
Use man to read about the find function
#Looking at find
man find
Use find to get a list from everything stored in the
/nfs/teaching/551-0132-00L/1_Unix1/genomes directory
# Getting a list with find
find /nfs/teaching/551-0132-00L/1_Unix1/genomes/
Use find to look for all .faa files there
# Looking fore .faa files
find . -name "*.faa"
Use find to look for all files larger than 5MB
# Looking fore files lager than 5MB
find . -size +5M
Now combine these criteria to find all .faa files larger than 5MB
# Looking fore .faa files larger than 5MB
find . -name "*.faa" -size +5M
Searching in less¶
When you open a file to look at it using less, it is also possible to search within that file by pressing / (search forwards) or ? (search backwards) followed by a pattern.
# Finding strings
/AAAA # finds the next instance of "AAAA"
?TTTT # finds the previous instance of "TTTT"
These same commands will also work with man, helping you to find a particular argument more easily.
But what happens when you search for “.”? The entire document will be highlighted! Why is this?
Regular Expressions¶
The reason this happens is that in the context of these search functions, “.” represents any character. It is acting as a wildcard, from a different set of wildcards to those discussed in Unix1.
This set of wildcards is part of a system of defining a search pattern called regular expression or regex. Such a pattern can consist of wildcards, groups and quantifiers, and may involve some complex logic which we will not cover here. Further, the exact set of wildcards available depends on the programming language being used.
# Wildcards and quantifiers
. any character
\d any digit
\w any letter or digit
\s any whitespace
^ the start of the string
$ the end of the string
* pattern is seen 0 or more times
+ pattern is seen 1 or more times
? pattern is seen 0 or 1 times
These are just a few of the possibilities available. An example regular expression that would search for email addresses, for instance, would be:
# name@domain.net can be matched as: \w+@\w+\.\w+
Let’s break this down:
The first part
\w+looks for any letter or digits one or more times, i.e.: the name part of the email address. Note that w does not match punctuation like “.” but does match underscores “_”.Then we ask for an at symbol
@.The second part
\w+again matches any alphanumeric string, i.e.: the domain part of the email address.Then we ask for an explicit full stop
\.which has to be delimited because a normal “.” matches any character.The third and final part is the same as the first and second and should match the net part of the email address.
So this is not a perfect regex for all email addresses because they can contain full stops and have more complex domain addresses.
Instead of searching for a regular expression describing a class such as w standing for any letter or digit, you could search for a specific expression such as the sequence ACGT. Enclosing this expression in brackets () turns it into a group. You can then also search for multiple occurences of this group by using brackets ()+ or you can simultaneously search for multiple patterns using the pipe character |. The pipe character acts as a logical OR operation here and divides the individual expressions within the group into alternates.
# Multiple occurences
ACGT would match ACGT
(ACGT)+ would match ACGT, ACGTACGT, ACGTACGTACGT etc.
(AC|CG|GT) would match AC, CG, GT
Grep¶
The command grep allows you to search within files without opening them first with another program. It also uses regular expressions to allow for powerful searches, and has a number of useful options to help give you the right output.
# A simple **grep**
grep "AAAAAAAAA" E.coli.fna # shows all lines containing "AAAAAAAAA" highlighted
# Using grep with a regex
grep -E "(ACGT)(ACGT)+" E.coli.fna # shows all lines containing "ACGTACGT.." highlighted
# Useful options
grep -o # show only the matches
grep -c # show only a count of the matches
Exercise 2.2
Navigate to the directory you copied the
E. colifiles to earlier.
# Navigation
cd ~/ecoli
Use less to look at the
GCF_000005845.2_ASM584v2_cds_from_genomic.fnafile, containing nucleotide gene sequences.
# Look at the file
less GCF_000005845.2_ASM584v2_cds_from_genomic.fna
Search within less to find the sequence for dnaA.
# Type this within less:
/dnaA
# Type 'n' or 'N' after to see if there are more search hits
# Press q to quit
Use man to look at the grep command
#Looking at grep
man grep
Use grep to find the same entry in the file.
#Using grep to search for dnaA
grep 'dnaA' GCF_000005845.2_ASM584v2_cds_from_genomic.fna
Use grep to count how many fasta entries the file has. As a reminder, a FASTA header always starts with a ‘>’.
# Use grep to count
grep -c '>' GCF_000005845.2_ASM584v2_cds_from_genomic.fna
Find the locus tag for the gene dnaA?
# Which entry?
grep '>.*dnaA.*' GCF_000005845.2_ASM584v2_cds_from_genomic.fna
If you are interested in learning regular expressions, try the exercises here
Data wrangling¶
A lot of time and effort in bioinformatics is spent arranging data in the correct way or correct format (aka “data wrangling”). Consequently, it is very useful to know how to filter and rearrange data files. In these exercises, we will learn some of the commands we use to do this.
The command sort will sort each line of a file, alphabetically by default, but other options are available.
# Sort some example files
cat /nfs/teaching/551-0132-00L/2_Unix2/sort_words.txt
sort /nfs/teaching/551-0132-00L/2_Unix2/sort_words.txt
#Sorting nummerically with the -n option
cat /nfs/teaching/551-0132-00L/2_Unix2/sort_nums.txt
sort -n /nfs/teaching/551-0132-00L/2_Unix2/sort_nums.txt
The command cut allows you to extract a single column of data from a file, for instance a .csv or .tsv file. The parameter -f describes which column will be extracted and can also be used to extract multiple columns.
# Look at some experimental metadata and extract the column we are interested in
less /nfs/teaching/551-0132-00L/2_Unix2/metadata.tsv
# Extract the 4th column from left to right
cut -f 4 /nfs/teaching/551-0132-00L/2_Unix2/metadata.tsv
# Extract multiple columns
cut -f 4,5 /nfs/teaching/551-0132-00L/2_Unix2/metadata.tsv
The command paste allows you to put data from different files into columns of the same file.
# Put together two files into one
paste /nfs/teaching/551-0132-00L/2_Unix2/sort_words.txt /nfs/teaching/551-0132-00L/2_Unix2/sort_nums.txt
The command tr will replace a given character set with another character set, but to use it properly you need to know how to combine commands (below).
# For instance, this command requires you to type the input in
tr 'ABCDEFGHIJKLMNOPQRSTUVWXYZ' 'abcdefghijklmnopqrstuvwxyz'
# Then try typing AN UPPER CASE SENTENCE
# Remember to exit a program that is running use ctrl + c
# It can also be used to delete characters
tr -d 'a'
# Then try typing a sentence with the letter 'a' in it.
# Remember to exit a program that is running use ctrl + c
The command uniq compresses adjacent repeated lines into one line, and is best used with sort when combining commands (see below).
# Look at a file and remove adjacent repeated lines
less /nfs/teaching/551-0132-00L/2_Unix2/uniq_nums.txt
uniq /nfs/teaching/551-0132-00L/2_Unix2/uniq_nums.txt
# Count how many times each value is repeated
uniq -c /nfs/teaching/551-0132-00L/2_Unix2/uniq_nums.txt
Exercise 2.3
Use the sort examples above and see what happens when you try to sort the sort_nums.txt file without the -n flag.
# Sort sort_nums.text without -n
sort /nfs/teaching/551-0132-00L/2_Unix2/sort_nums.txt
# The file will be sorted alphabetically
Look at the file
/nfs/teaching/551-0132-00L/2_Unix2/sort_tab.txt.
# Look at sort_tab.txt
less /nfs/teaching/551-0132-00L/2_Unix2/sort_tab.txt
Extract the second column of this file using cut.
# Extract the second column
cut -f 2 /nfs/teaching/551-0132-00L/2_Unix2/sort_tab.txt
Looking at the manual for sort, can you figure out how to sort sort_tab.txt according to the second column, or ‘key’?
# Looking at he manual
man sort
# Sort the table by second column
sort -n -k 2 /nfs/teaching/551-0132-00L/2_Unix2/sort_tab.txt
# Note that if you forget the -n then the numbers are sorted alphabetically, not numerically
Use paste to combine the two files sort_words.txt and sort_nums.txt (in the directory
/nfs/teaching/551-0132-00L/2_Unix2/) into a single two-column output.
# Use paste to combine files
paste /nfs/teaching/551-0132-00L/2_Unix2/sort_words.txt /nfs/teaching/551-0132-00L/2_Unix2/sort_nums.txt
Use tr so that when you enter the word banana it comes out as rococo.
# Use tr to convert one word into another
tr 'ban' 'roc'
# Then input banana and back comes rococo!
# Use ctr + c to kill the command
Use the uniq examples above, then check with uniq -c that each line in sort_tab.txt is unique.
# Check file with uniq
uniq -c /nfs/teaching/551-0132-00L/2_Unix2/sort_tab.txt
# Each value in the first column is 1 - no repeats!
Combining commands¶
The power of this set of commands comes when you use them together, and when you can save your manipulated data into a file. To understand how to do this we have to think about the command line input and output data.
Input and output¶
So far we have been using files as arguments for the commands we have practiced. The computer looks at the memory where the file is stored and then passes it through RAM to the processor, where it can perform whatever you have asked it to. We have seen output on the terminal, but it’s equally possible to store that output in memory, as a file. Similarly, if we want to use the output of one command as the input to a second command, we can bypass the step where we make an intermediate file.
The command line understands this in terms of data streams, which are communication channels you can direct to/from files or further commands:

stdin: the standard data input stream
stdout: the standard data output stream (defaults to appearing on the terminal)
stderr: the standard error stream (also defaults to the terminal)
Although you can usually give files as input to a program through an argument, you can also use stdin. Further, you can redirect the output of stdout and stderr to files of your choice.
# Copy and rename the file containing the E.coli genome
cd
cp /nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.fna E.coli.fna
# Using the standard streams
head < E.coli.fna # send the file to head via stdin using '<'
head E.coli.fna > E.coli_head.fna # send stdout to a new file using '>'
head E.coli.fna 2> E.coli_err.fna # send stderr to a new file using '2>'
head E.coli.fna &> Ecoli_both.fna # send both stdout and stderr to the same file using '&>'
head < E.coli.fna > E.coli_head.fna 2> E.coli_err.fna # combine all three stdin, stdout, stderr
Chaining programs together¶
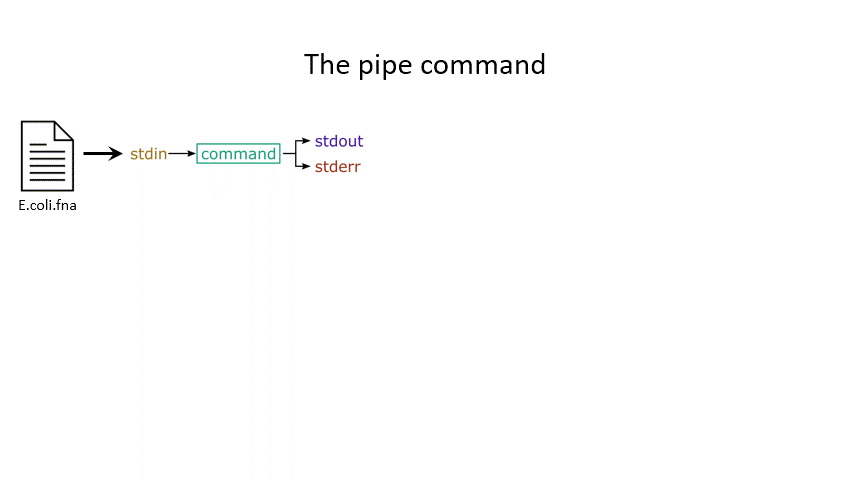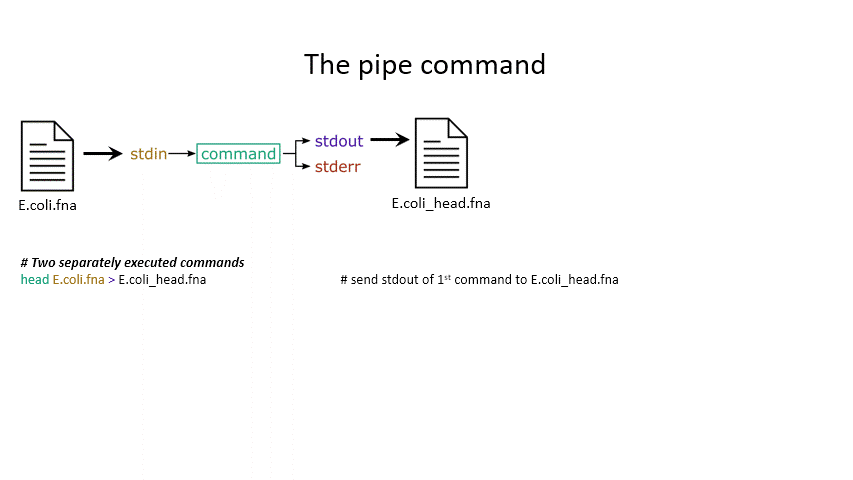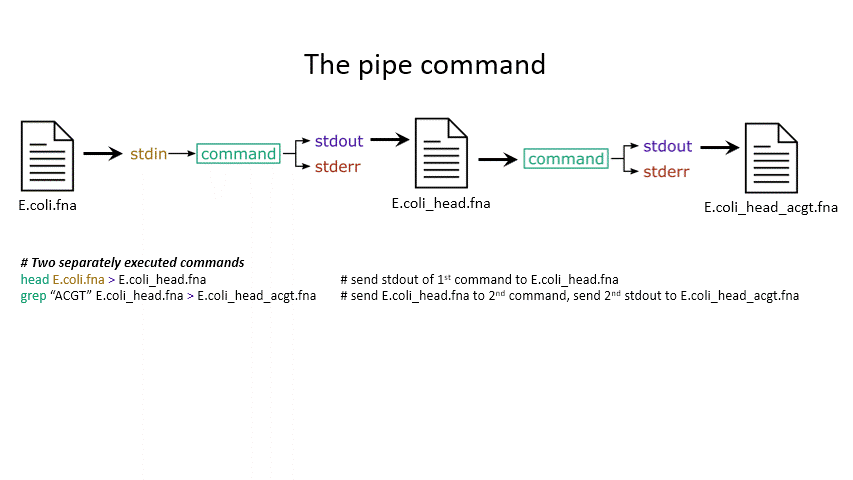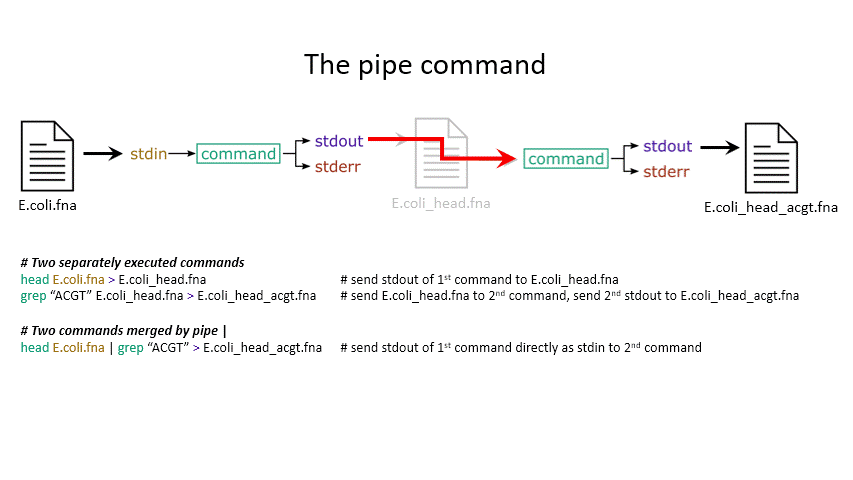
Sometimes you want to take the output of one program and use it as the input for another – for instance, run grep on only the first 10 lines of a file from head. This is a procedure known as piping and requires you to put the | character in between commands (although this may not work with more complex programs).
# Copy and rename the file containing the E.coli open reading frames
cd
cp /nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna E.coli_CDS.fna
# Piping
head E.coli.fna | grep "ACGT" # send the output of head to grep and search
grep -A 1 ">" E.coli_CDS.fna | grep -c "^ATG" # use grep to find the first line of sequence of each gene and send it to a second grep to see if the gene starts with ATG
Exercise 2.4
Rename the file GCF_000005845.2_ASM584v2_cds_from_genomic.fna (put in your home directory earlier) to E.coli_CDS.fna
# Copy the file to your home directory
cp /nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna ~/E.coli_CDS.fna
Use grep to find all the fasta headers in this file, remember that a fasta header line starts with ‘>’.
# Find the fasta headers
grep '^>' E.coli_CDS.fna
Send the output of this search to a new file called cds_headers.txt.
# Send the output to a new file
grep '^>' E.coli_CDS.fna > cds_headers.txt
Use grep again to find only the headers with gene name information, which looks like, for instance [gene=lacZ], and save the results in another new file called named_cds.txt.
# Find named genes
grep '\[gene=' cds_headers.txt > named_cds.txt
Use wc to count how many lines are in the file you made.
# Count how many there are
wc -l named_cds.txt
Now repeat this exercise without making the intermediate files, instead using pipes.
# Repeat without intermediate files
grep '^>' E.coli_CDS.fna | grep '\[gene=' | wc -l
As an additional challenge:
Using the commands we have used, find the start codon of each gene in E. coli and then count up the frequency of the different start codons.
# Count the frequency of start codons in the *E.coli* genome
grep -A 1 '^>' E.coli_CDS.fna | grep -Eo '^[ACGT]{3}' | sort | uniq -c | sort -nr -k 1
# The first part finds all headers plus the first line of sequence
# The second part is a regular expression to find the first three nucleotides in the sequence lines
# Then we have to sort them so that we can count them with uniq
# The final part is a bonus that sorts by descending frequency
# And as so often in bioinformatics, there are several ways of getting a task done.
# Consider the following alternative:
grep -A 1 ">" E.coli_CDS.fna | grep -v '>' | grep -o "^\w\w\w" | sort | uniq -c | sort -k1nr
Writing and running a script¶
If you construct a series of commands that you want to perform repeatedly, you can write them into a script and then run this script instead of each command individually. This makes it less likely that you make an error in one of the individual commands, and also keeps a record of the computation you performed so that your work is reproducible.
You can write the script using a text editor on your computer and then uploading it with scp (for examble notepad). If you want to write a script directly in the terminal there are text editors available such as vim and emacs - you should be able to find tutorials for both online (for example openvim or GNU Emacs).
By convention, a script should be named ending in .sh and is run as follows:
# Run a script in the same directory
./myscript.sh
# Run a script in another directory
./mydir/myscript.sh
The command line interface, or shell, that we use is called bash and it allows you to execute the same script with changing arguments, encoded as variables $1, $2, etc.
For instance we could have a simple script:
# myscript.sh
echo "Hello, my name is $1"
# Running my simple script
./myscript.sh Chris
"Hello, my name is Chris"
This means you could write a script that performs some operations on a file, and then replace the file path in your code with $1 to allow you to declare the file when you execute the script. Just remember that if your script changes working directory, the relative path to your file may be incorrect, so sometimes it is best to use the absolute path.
File permissions¶
A very useful option for the command ls is -l, often given as alias ll. So if you use ll instead of ls it will not only list the file names within a directory but also give you information about file permissions, owner, file size, modification date etc.
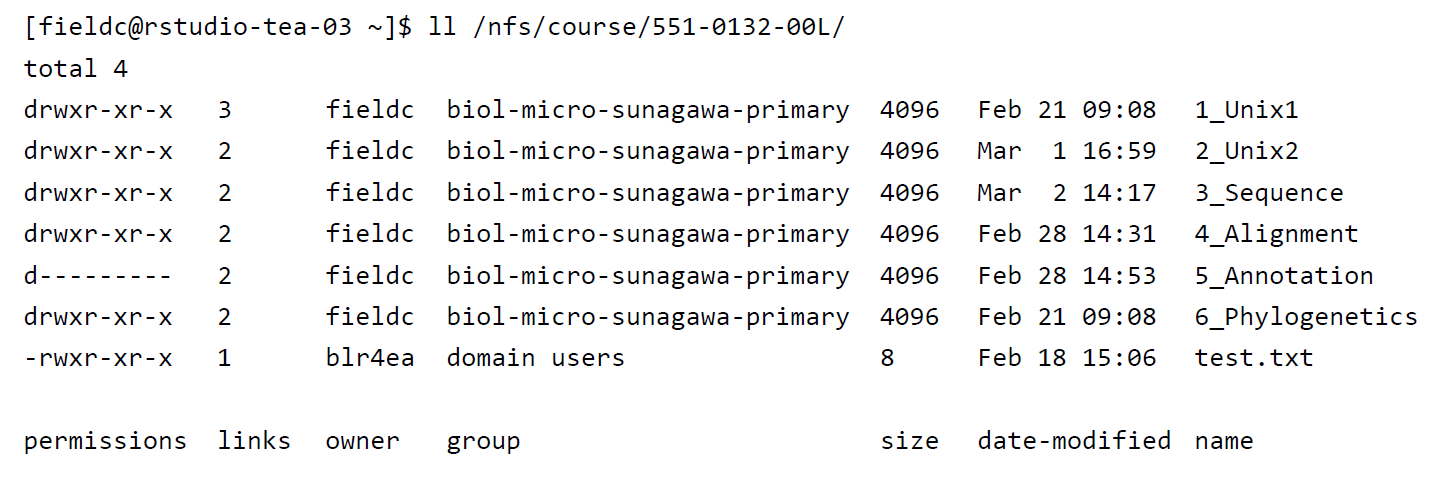
File permissions are useful to define who has access to read a file, write (make changes to the file) and execute (run a script) a file. Permissions are always split into 3 sections, the first section describing the permissions for the file owner, the second section describing permissions for a defined user group and the third section describing permissions for everyone else. Within each section, permissions are split into read r, write/edit w and execute/run x permissions.

# File permissions
drwxrwxrwx Any user (owner, user group, everyone) has permissions to read, write and execute a file
drwxr-xr-x The owner has permission to read, write, execute, while the user group and everyone only have read, execute permissions
drwxrwx--- The owner and user group have access to read, write and execute the file while others cannot access the file at all
Exercise 2.5
Write a simple script that will count the number of entries in a fasta file
Use a variable to allow you to declare the file when you run the script
# Use the text editor of your choice. Here we are going to use vim.
vim fastacount.sh
# Press "i" to enable the writing option and write a simple script to count fasta entries in a file.
grep -c "^>" $1
# You can exit your script by pressin "esc" and typing ":wq" (w for saving and q for quitting).
Make your script executable with the command “chmod +x”
# Make it executable
chmod +x fastacount.sh
Test it on some of the fasta files in the
/nfs/teaching/551-0132-00L/1_Unix1/genomessubdirectories
# Run the script
./fastacount.sh /nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna # 4302
Software¶
Whenever you want to perform more complex computational tasks, you might use already existing pieces of software. These are essentially bundles of scripts carrying out a series of computational tasks with many dependencies. In order to install these pieces of software, you could (from easiest to hardest):
Ask your friendly sysadmin to install it for you
Use a docker or singularity image
Install it using Conda
Install it from scratch, including libraries, dependencies and ensuring that it does not clash with any existing installations
As these installation processes can be time consuming and tedious, we have a well-maintained module system with pre-installed and continuously updated versions of software which is commonly used. This pre-installed software automatically loads the required libraries and dependencies and thereby avoids clashes.
# Commands for using the module system
ml or module base command; list loaded modules
ml avail list available modules
ml <name> or ml load <name> load named module
ml unload <name> unload named module; after use
ml purge unload all loaded modules
ml spider <name> search modules for name
Working on a computing cluster¶
The Slurm Queuing System¶
Many people have access to Euler. If everyone ran whatever program they liked, whenever they liked, the system would soon grind to a halt as it tried to manage the limited resources between all the users. To prevent this, and to ensure fair usage of the server, there is a queueing system that automatically manages which jobs are run when. Any program that will use either more than 1 CPUs (sometimes referred to as cores or threads, though there are minor technical differences between these terms), more than a few MB of RAM, or will run for longer than a few minutes, should be placed in the queue.
To correctly submit a job to the queue on Euler, it’s usually easiest to write a short shell script based on a template. Our server Cousteau also uses the Slurm Queuing System.
#! /bin/bash
#SBATCH --job-name example # Job name
#SBATCH --output example_out.log # Output log file path
#SBATCH --error example_error.log # Error log file path
#SBATCH --ntasks 8 # Number of CPUs
#SBATCH --mem-per-cpu=2G # Memory per CPU
#SBATCH --time=1:00:00 # Approximate time needed
# Insert your commands here
echo This job ran with $SLURM_NTASKS threads on $SLURM_JOB_NODELIST
Then the equivalent commands:
# Submit the job to the queue
sbatch my_jobscript.sh
# Check the status of your jobs
squeue
# Remove a job from the queue
scancel jobid
Exercise 2.6
Copy the script
/nfs/teaching/551-0132-00L/2_Unix2/submit_slurm.shto your home directory.
# Copy the submit script to your home directory
cp /nfs/teaching/551-0132-00L/2_Unix2/submit_slurm.sh ~/
Submit the script to the job queue with sbatch and look at the output file
# Submit the script
sbatch submit_slurm.sh
# Check if it is in the queue (it may run too fast and you miss it)
squeue
# Check the output files
less example*error.txt # Note: "*" stands for a number
# Should be empty
less example*out.txt # Note: "*" stands for a number
# Should tell you that it ran with 8 threads on localhost
Now edit the script:
Remove the existing echo command.
Put a command to run the script you wrote for Exercise 2.5 on one of the fasta files in
/nfs/teaching/551-0132-00L/1_Unix1/genomes.You should only use 1 CPU instead of 8, the other parameters can stay the same unless you want to rename the job and log files
# Modify the submit script (submit_slurm.sh) to look something like this:
#! /bin/bash
#SBATCH --job-name fastacount # Job name
#SBATCH --output out.log # Output log file path
#SBATCH --error error.log # Error log file path
#SBATCH --ntasks 1 # Number of CPUs
#SBATCH --mem-per-cpu=2G # Memory per CPU
#SBATCH --time=1:00:00 # Approximate time needed
./fastacount.sh /nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_cds_from_genomic.fna
Submit the job. When the job is finished, look at the output files for yourself.
# Then you submit it like this:
sbatch submit_slurm.sh
# Check the output
less error.log # Should be empty
less out.log # Should have the output of your script, e.g. 4302