Alignment¶
General information¶
Topic overview¶
In this lesson you will learn about the concept of sequence alignment. You will learn how to use BLAST via the web-based interface from the NCBI and via the command line. You will also learn how to perform multiple sequence alignment.
Learning objectives¶
Learning objective 1 - You are able to use the BLAST software to align sequences against one another and search for similarity in sequence databases
You use the BLAST web interface to perform alignment and search
You run BLAST algorithms on the command line to perform alignment and search
Learning objective 2 - You comprehend the challenges of multiple sequence alignment and recognise different approaches to the problem
You perform multiple sequence alignment on the command line with software such as MUSCLE
You inspect the output of multiple alignment and interpret the quality
Comparative Sequence Analysis¶
In this section of the course, you will learn how to analyse an unknown sequence. For simplicity, we will focus on the procedure you would follow for the genes in a novel bacterial genome. The techniques we use are generally applicable to other organisms and other features, but more difficult or computationally intensive. We will briefly discuss other applications beyond bacterial genes in the relevant sections.
The common element in all these techniques is sequence comparison. Determining the similarities and differences between two or more sequences allows us to infer the features and functions of the sequence (annotation) and its relationship to other sequences (phylogeny). However in order to compare sequences we must understand the concept of alignment.
Sequence alignment¶
One way to compare two sequences is through alignment - arranging them against one another to identify areas of similarity. There are two general approaches you could take:
Global alignment attempts to align every residue (every base in a nucleotide sequence, every amino acid in a protein sequence) between the two sequences
Local alignment looks for regions of alignment between the two sequences and ignores the rest (dissimilar regions)
We then need an algorithm that will arrange and score different possible alignments.
BLAST¶
BLAST, or the Basic Local Alignment Search Tool (BLAST), is the most commonly used alignment tool and, which you may be able to tell from its acronym, performs local alignment. The details of the algorithm (here) are beyond the scope of this course, but understanding the scoring system is important.
BLAST scores an alignment residue by residue based on whether it is a match, mismatch, or a gap (which might exist due to the insertion or deletion of 1 or more residues in one sequence).
mismatch
|
ATGACTAGCTGCTATATCAGCTAC
|| |||||||| ||||||| <-- every | indicates a match
GTG-CTAGCTGC-----CAGCTAC
| |
gap extended gap
The score for a match or mismatch depends on exactly which residues are being compared. For DNA this is simple, an identical penalty for any mismatch. For amino acids this is more complex, where residues with similar properties (say, hydrophobicity or polarity) are penalised less than those that are very different. A scoring matrix is employed to determine the final match or mismatch score and here we show the BLAST DNA scoring matrix and a commonly used amino acid matrix, BLOSUM62:
A |
C |
G |
T |
|
A |
+5 |
-4 |
-4 |
-4 |
C |
-4 |
+5 |
-4 |
-4 |
G |
-4 |
-4 |
+5 |
-4 |
T |
-4 |
-4 |
-4 |
+5 |

Gaps are scored in two ways. Firstly, there is a penalty for opening a gap, and if there are multiple gaps in a row then a gap extension penalty is applied, typically less than the penalty for opening it in the first place. In BLAST, these penalties depend on the exact algorithm used.
So the final score for a given alignment is the sum across all residues of the matches, mismatches, gap openings and gap extensions.
Using BLAST online¶
Most researchers are familiar with the web-based interface for BLAST found here.
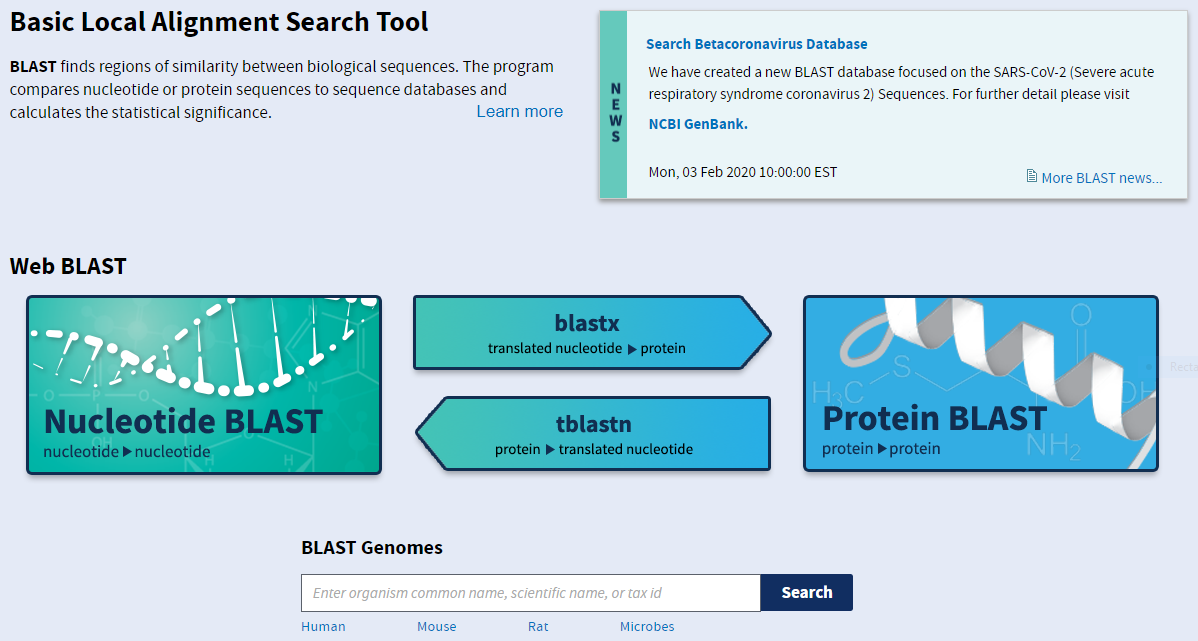
The four main varieties of BLAST are neatly summarised on this frontpage.
blastn: nucleotide blast for comparing DNA sequences (left)
blastx: searching a protein database for a translated nucleotide query (middle top)
tblastn: searching a translated nucleotide database for a protein query (middle bottom)
blastp: protein blast for comparing protein sequences (right)
You should therefore choose your BLAST algorithm based on the nature, nucleotide or protein, of your query and the database against which you want to compare it. There are also further versions available for more specialised applications.
More details about BLAST on the NCBI-Webpage can be found here.
Pairwise alignment¶
We will first select Nucleotide BLAST (blastn) to perform a pairwise alignment of two nucleotide sequences that we have put in the files /nfs/teaching/551-0132-00L/4_Alignment/pairwise1.fasta and /nfs/teaching/551-0132-00L/4_Alignment/pairwise2.fasta.
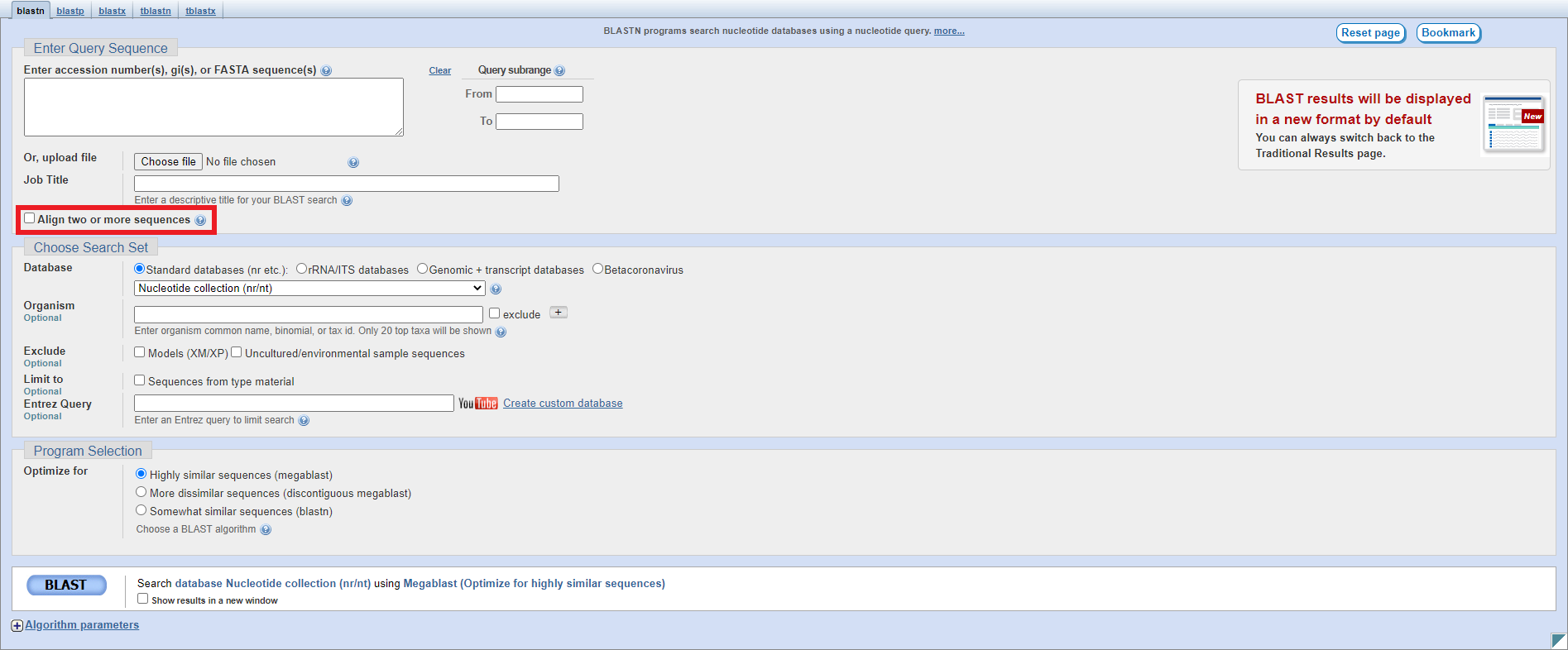
The setup page for blastn looks as follows - you should click on the checkbox highlighted to enable pairwise alignment:
This will change the page to look as follows:
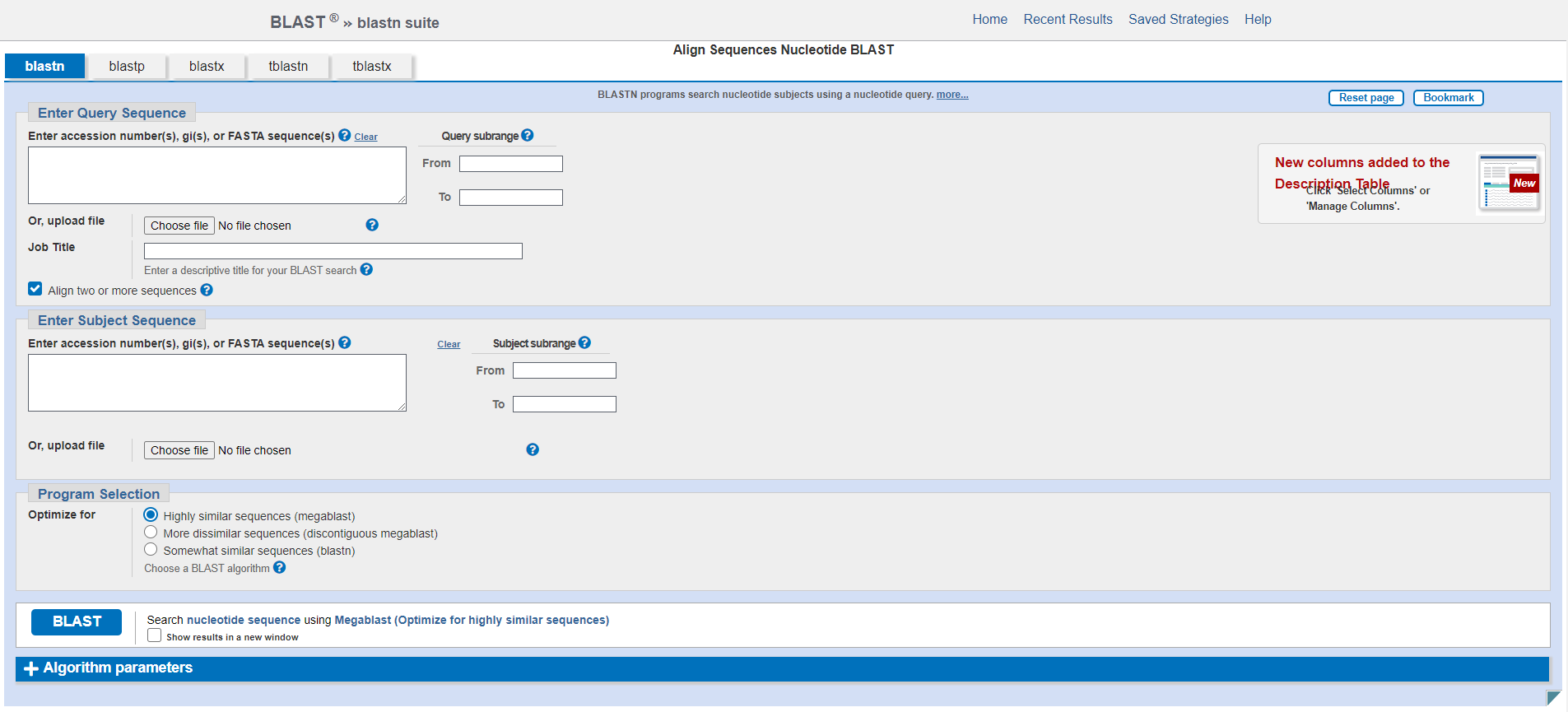
In each of the sections Enter Query Sequence and Enter Subject Sequence you can either paste the relevant sequence into the text box directly or choose a file to upload, which should be in fasta format. For each sequence you can also specify a subrange from the sequence by giving start and end coordinates. You can also give your alignment a Job Title.
The Program Selection section allows you to select which specific blastn algorithm you want to use. Depending on your application and more specifically the expected sequence similarity you may want to choose a different program to optimize your search and algorithm speed. The default program megablast is very fast and works best for highly similar sequences (with expected sequence percent identity >= 95%). If you expect your sequences to be a bit more dissimilar, you might want to use discontiguous blast which allows for more mismatches. blastn is the slowest alorithm but allows much shorter alignments and therefore enables to retrieve alignments between somewhat similar (more distant) sequences.
Exercise 4.1
Perform a pairwise nucleotide BLAST alignment of the sequences in the files
/nfs/teaching/551-0132-00L/4_Alignment/pairwise1.fastaand/nfs/teaching/551-0132-00L/4_Alignment/pairwise2.fasta. For this example, you don’t need to enter any subrange coordinates or change the algorithm from the default (megablast). It is up to you whether you want to copy and paste the sequences or upload the fasta files.
# Make sure the "Align two or more sequences" box is ticked
# Open the file pairwise1.fasta with less and copy the nucleotide sequence and paste under "Enter Query Sequence"
less /nfs/teaching/551-0132-00L/4_Alignment/pairwise1.fasta
# Repeat for pairwise2.fasta but paste the sequence under "Enter Subject Sequence"
less /nfs/teaching/551-0132-00L/4_Alignment/pairwise2.fasta
# You can also download the files first with the scp command you learned in Unix1 and upload the files to NCBI
As we learned previously, megablast works well for sequences which are highly similar and is very fast, so in this case we can use megablast because we know that the two example sequences are highly similar
Investigate the result by reading through the “Descriptions” tab, specifically the parameters Max Score, Query Coverage and Percent Identity
Check the alignment in detail by clicking on the tab “Alignments”
Alignment results¶
The results page has a summary of the search performed at the top (left), with the option to further filter the results (right).
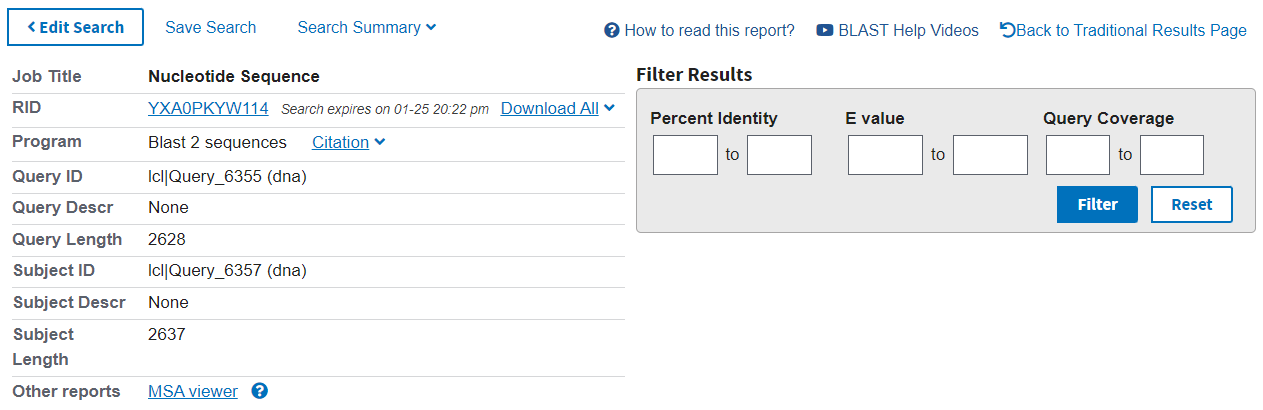
The first tab in the results section is Description. Here you can see the statistics for each alignment found between the two sequences:
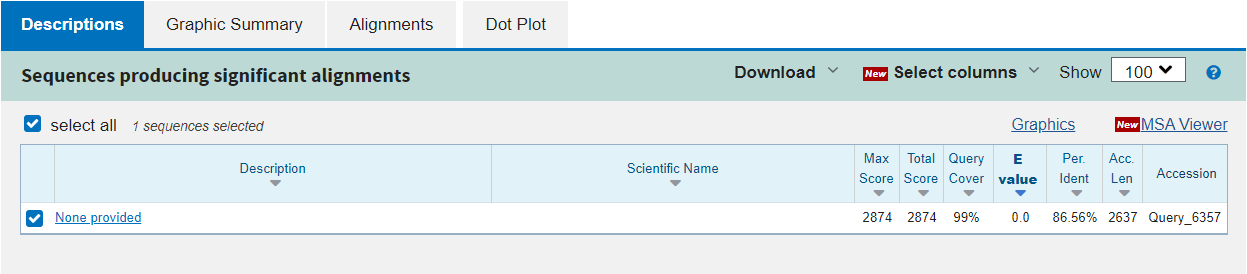
Max Score: the highest score from the alignments found
Total Score: the sum of scores for all alignments found
Query Cover: the percentage of the query sequence for which any alignment was found
E value: the likelihood of the alignment being seen by chance (note that this is dependent on the database size searched)
Per. Ident: the percentage identity of the alignment, i.e.: how many base pairs are identical
Acc. Len: the length of the subject sequence
Accession: the name of the subject sequence (or an arbitrary name)
In this example, there is only one alignment in the results and so some of this information is not interesting. What we can see is that the alignment covers 99% of our query sequence and is approximately 87% identical. We cannot really say if this alignment is significant or not because we have only compared our query to one subject, and this was contrived to give a successful alignment. Nonetheless we can inspect the alignment more carefully in the other tabs.
Warning
The ‘percent identity’ of an alignment is sometimes, perhaps misleadingly, referred to as ‘percent homology’ - however, this implies a shared evolutionary history. As two sequences that perform the same function could well have evolved independently, sequence similarity suggests but is not sufficient proof of homology.
The second tab gives a graphical summary of the alignment, depicting the quality across the length of the query sequence with a colour code:
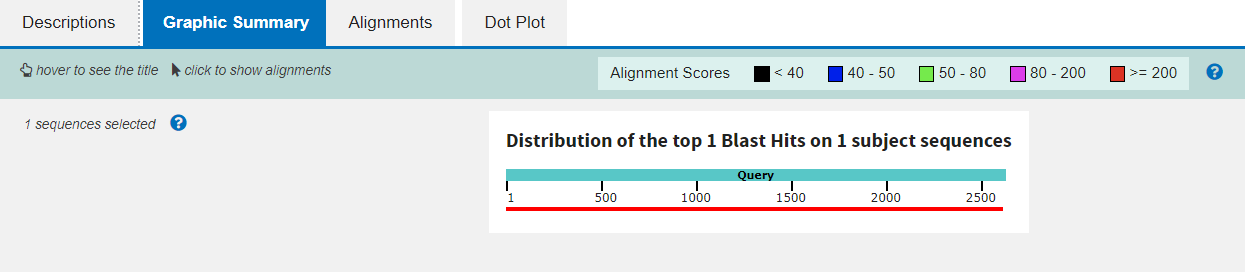
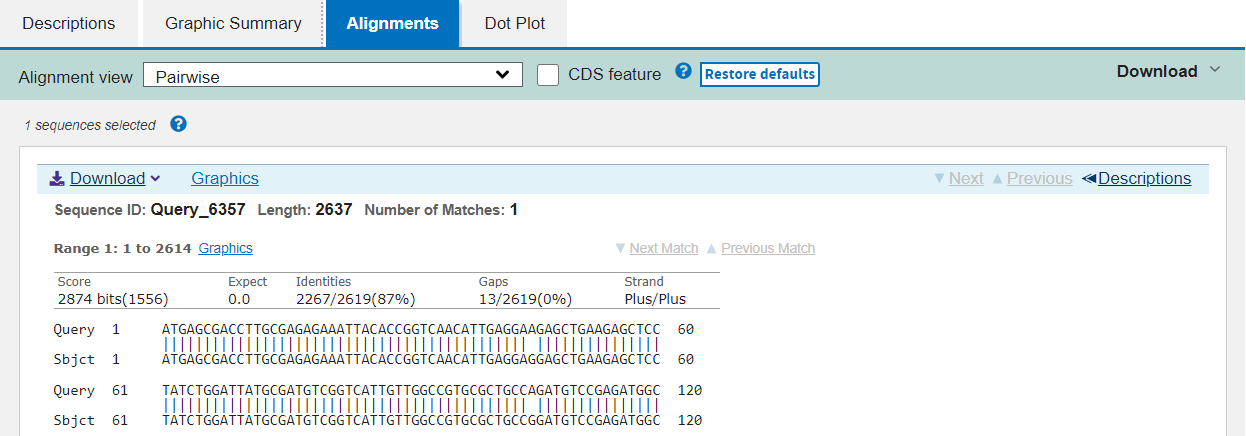
The third tab shows you the precise alignment summarised in the first tab. The query and subject sequences are shown beside one another, with vertical pipe symbols “|” representing identity and dashes “-” for gaps in either sequence.
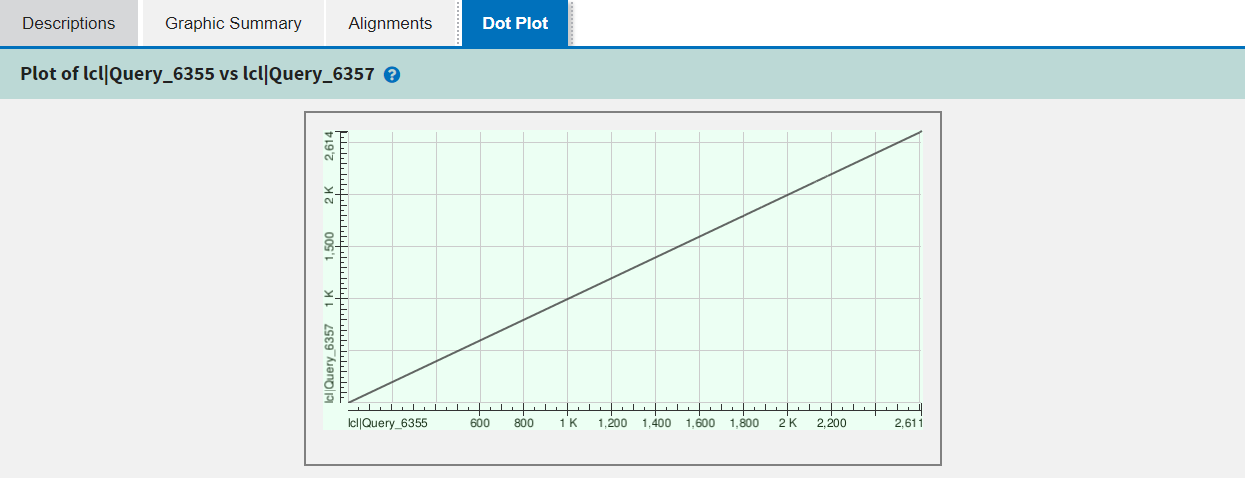
The fourth and final tab is specific to pairwise alignment and shows a dot plot of the alignment or alignments between the pair of sequences.
Exercise 4.2
For the alignment you just performed, what would happen if you enter subrange coordinates from 1 to 600 for pairwise1.fasta and from 500 to 1000 for pairwise2.fasta? You might have an idea based on your previous result, but if you are not sure you can simply try it out.
Repeat the alignment with 100 bases overlapping subrange coordinates for the two sequences
You will see that there is no longer an alignment between the two sequences even if we know that they align along the whole sequence length and also along the subrange overlap
This is important to realize because subrange coordinates should be used with care and only if 1) you are certain that there is no similarity in the disregarded ranges or 2) you are only interested in alignments within a certain range
What would happen if you extend the subrange coordinates from 1 to 750 for pairwise1.fasta and you keep the subrange from 500 to 1000 for pairwise2.fasta? If you are not sure you can simply try it out.
Repeat the alignment with a bigger overlap between subrange coordinates for the two sequences
Extending the subrange overlap allows you to recover the sequence alignment
Depending on the number of mismatches and gaps the total alignment score will be impacted so that the sequence alignment needs to be a certain length to show up as alignment
You can check the tab “Alignments” again and will see that only the subrange overlap shows up because BLAST is a local alignment algorithm and thus the rest of the sequences that does not align is ignored
Can you think of a different way to retrieve the alignment between the subrange 1 to 600 for pairwise1.fasta and the subrange 500 to 1000 for pairwise2.fasta? Hint: If we do not change the chosen coordinate subranges and sequences, the retrieval of the alignment will be directly dependent on the scores/penalties of matches/mismatches/gaps defined by the alignment algorithm.
As we learned previously, megablast works well for sequences which are highly similar and is very fast
If we do not know how similar/distant two sequences are, we would rather use the discontiguous megablast or blastn algorithm which are slower but can retrieve more distantly related sequences
If we select either of the slower algorithms we can retrieve the alignment of 100 bases alignment between the two sequences that we could not retrieve with megablast
However is important to note that these other algorithms are much slower and take longer until they display a results
Searching by alignment¶
BLAST is used far more often for searching by alignment, that is, finding sequences in a database that align to a query (i.e. your sequence of interest). If we return to the BLAST homepage and again select Nucleotide BLAST (blastn), and then make sure that the “Align two or more sequences” is unchecked, we should see the standard search interface:
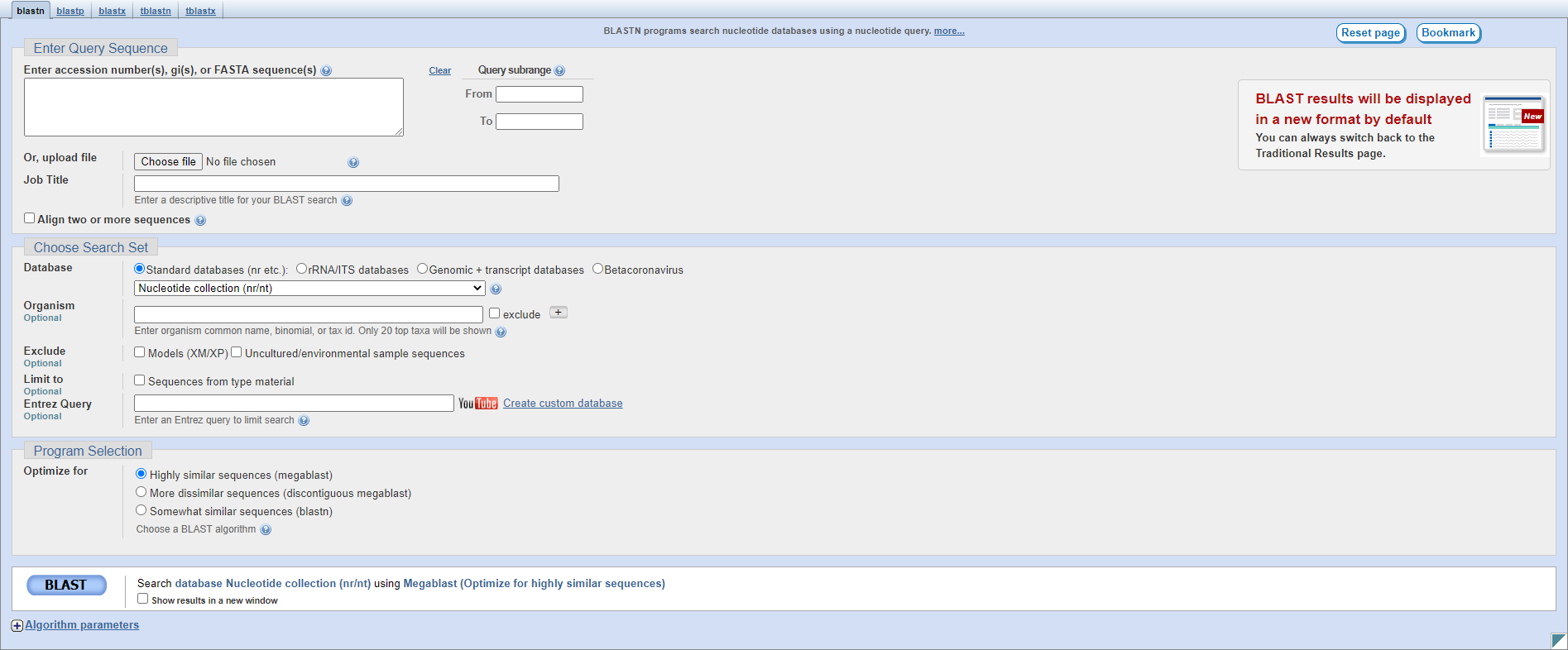
Let’s look at the different sections on this page:
Enter Query Sequence: this section is the same as for pairwise alignment.
Choose Search Set: here you can specify search parameters.
Database: this is where you select an NCBI database to search; typically nr/nt to search Genbank protein/nucleotide sequences but you may want to use RefSeq or another specific database.
Organism: you can enter the name of an organism or taxonomic level to restrict your search, for instance Escherichia or fungi.
Exclude: checkboxes to exclude RNA and protein sequences generated by NCBI’s automated pipeline (the Models), or uncultured sequences (those reconstructed from metagenomic samples).
Limit to: you can restrict your search to only type material, which are the exemplary species, see this paper for more information.
Entrez Query: you can also use custom search terms from the Entrez system to restrict your search.
Program Selection: you can select the specific algorithm you want to use here; the default is usually sufficient but you can use a slower but more sensitive algorithm if needed.
BLAST: this button will begin the search.
Algorithm parameters: here you can specify the exact parameters BLAST will use in alignment during the search; in general you won’t need to modify those used by the three Program Selection options.
Search results¶
The results page has a summary of the search performed at the top (left), with the option to further filter the results (right).
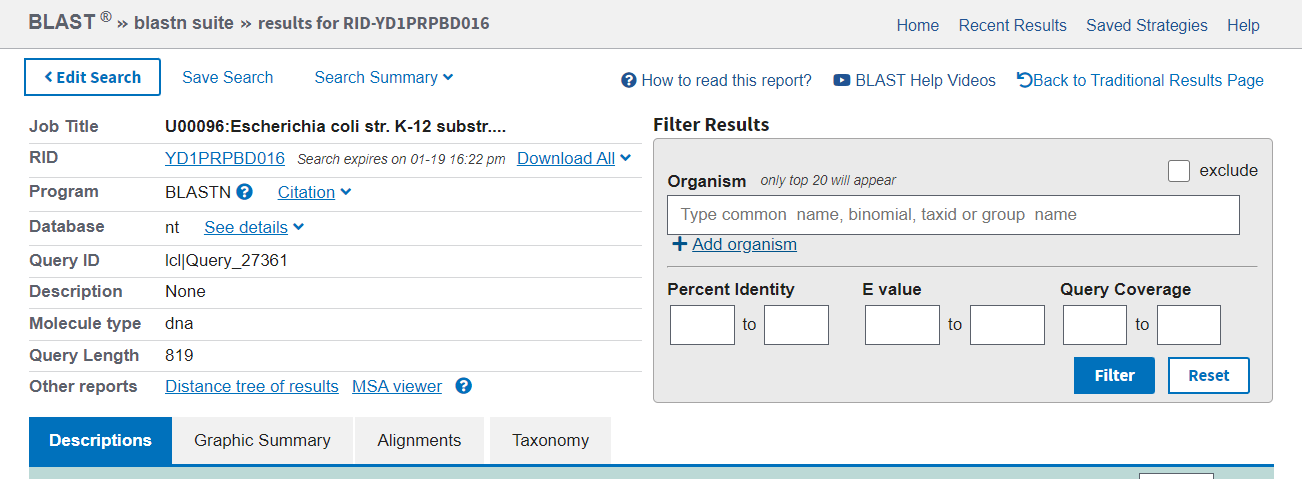
The first tab in the results section is Description. The statistics reported are the same as for pairwise alignment. You can sort hits by any of the data columns if you want to specifically find the longest or most identical hit, for instance, where the default is by e-value.
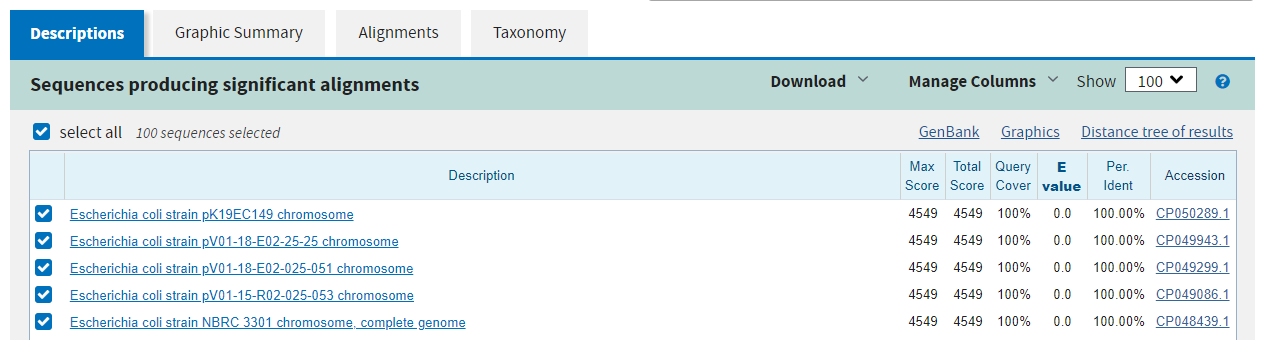
The second tab gives a graphical summary of the hits found. It depicts the quality of the alignment across the length of the query sequence with a colour code for alignment quality.

The third tab shows you the precise alignment found by the algorithm. The query and subject sequences are shown beside one another, with vertical pipe symbols “|” representing identity.
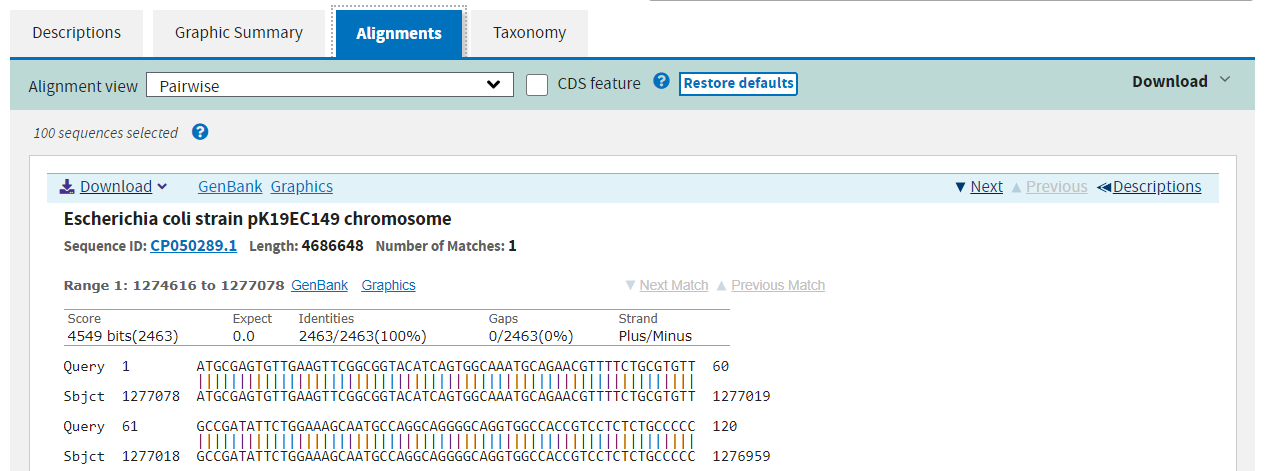
The fourth and final tab gives you a taxonomy of the organisms found in the hits and how many hits were found at each taxonomic level.
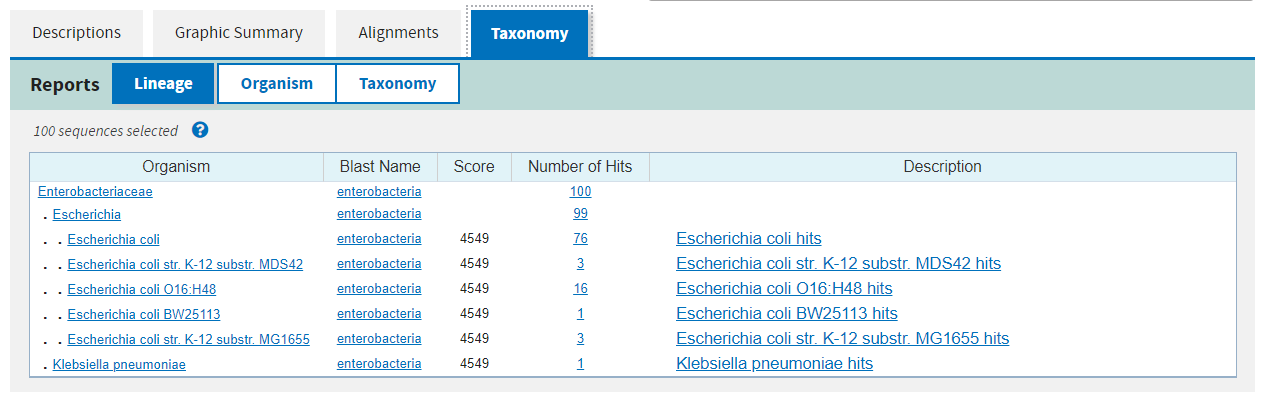
Exercise 4.3
Perform a nucleotide BLAST search for sequence found in the file
/nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_01.fasta
# Make sure the «Align two or more sequences» box is not ticked and the webpage has switched to the database search mode
# Open the mystery sequence and copy/paste it into the search field
less /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_01.fasta
Based on the results, what organism do you think the sequence is?
Based on the search you should find out that the sequence is from the Mycobacterium tuberculosis bacterium.
Based on that information, can you find a way to identify what protein the gene sequence encodes for?
Go back to the NCBI blast starting page and select blastx (translated nucleotide to protein)
Make sure the «Align two or more sequences» box is not ticked and the webpage has switched to the database search mode
Enter your sequence or upload the file mystery_sequence_01.fasta as query
Note: Since multiple genetic codes exists, you ideally know at least what group of organisms a sequence belongs to before you try to investigate the protein it encodes, you can then decide which genetic code to use (in this case choose Bacteria and Archaea (11))
Note: You can also specify the organism (if known) to further increase the efficiency of your search
The result (mystery_sequence_03.faa) shows that the DNA sequence encodes for DNA topoisomerase (ATP-hydrolizing) subunit A/DNA gyrase subunit A from Mycobacterium tuberculosis
If you now want to know how similar this protein sequence is to the same protein within another organism (mystery_sequence_04.faa), how would you do this?
Go back to the NCBI blast starting page and select blastp (protein-protein alignment)
Make sure the «Align two or more sequences» box is ticked
Copy/paste the protein sequence from the previous exercise in the first entry (click on Alignments subpage, click on first alignment, click on the sequence ID, click on FASTA, copy/paste) or copy/paste or upload from file mystery_sequence_03.faa
Open the file mystery_sequence_04.faa and copy/paste this protein sequence into the second entry (or upload the file)
Checking the alignment, you will see that there are 62 positions with differences in encoded amino acids
If you are instead interested in the total number of differences between the two genes encoding for the two proteins, you would want to compare them at the DNA level. Why is this the case and how would you do this?
Because of the redundancy of the genetic code, some triplets of DNA nucleobases encode for the same amino acid. Thus differences in DNA sequences leading to the same amino acid are not visible at the protein level.
Make sure the «Align two or more sequences» box is not ticked and the webpage has switched to the database search mode
Go back to the NCBI blast starting page and select tblastn (protein to translated nucleotide)
Enter the mystery_sequence_04.faa and retrieve the DNA sequence from the results
Note that the output of this search returns the complete genome sequence instead of only the gene (this does not matter much though because BLAST is a local alignment which will still find alignments also between sequences of very different length where most parts do not align)
Go back to the NCBI blast starting page and select blastn (nucleotide-nucleotide alignment)
Make sure the «Align two or more sequences» box is ticked
Enter the mystery_sequence_01.fasta as first entry
Enter the DNA sequence that you just translated from mystery_sequence_04.faa as a second entry (mystery_sequence_05.fasta)
Checking the alignment, you will see that there are 340 positions with differences in the DNA sequence
Note that the 62 amino acids could only lead to 3*62=186 changes in amino acids, thus the rest of the differences are only present at the DNA level but not at the protein level
BLAST on the command line¶
The various BLAST programs are also available for download to run yourself. The BLAST+ suite is installed on the server Cousteau and Euler. There are several reasons you might want to run BLAST offline:
You may have a large number of searches you want to perform, which is clumsy via the web interface
You have sensitive data you want to search for or with that you shouldn’t submit online
You want to perform BLAST searches as part of a larger program or software pipeline
Making a BLAST database¶
In order to run a search offline, you need a database to search. You can download one of the NCBI databases if you have the storage space, but hopefully your friendly local bioinformatician has already done that for you - and indeed we have made the RefSeq Prokaryotic Reference Genome database available here:
/nfs/teaching/databases/ncbi/ref_prok_rep_genomes
It’s also likely that you want to search a much smaller and specific set of sequences that you have already prepared in FASTA format. For this, there is the command makeblastdb (which is typically used as soon as you have more than 2 sequences to align):
# Make a nucleotide sequence database
makeblastdb -in dna_sequences_to_search.fasta -dbtype nucl
The -in argument should point to a fasta file that you want to create the database from, and the -dbtype must be either ‘nucl’ for nucleotide sequence or ‘prot’ for protein sequence. Other options allow you to use different types of input file, to mask sequences or parts of sequences, to index taxonomic information for the database and more. However you run it, makeblastdb produces some additional files if you check the directory containing the FASTA file that index the sequences ready for searching.
Running BLAST¶
The four BLAST algorithms highlighted on the front page of the NCBI BLAST homepage have identically-named command line equivalents. The software suite is available via our module system, called BLAST+. We will demonstrate here with blastn, and some of the arguments vary slightly for the other algorithms, but the most important thing is to choose the correct one based on the nature of your query and database (as described above).
Firstly, you can perform pairwise alignment just as with the online interface by specifying a query (sequence of interest) and subject (for comparison) sequence, as in this minimal example:
# Load the software module
ml BLAST+
# Pairwise alignment with blastn
blastn -query /nfs/teaching/551-0132-00L/4_Alignment/pairwise1.fasta -subject /nfs/teaching/551-0132-00L/4_Alignment/pairwise2.fasta
Secondly, you can of course search by alignment by providing a database to look through, as in this minimal example:
# Run blastn
blastn -db /path/to/db -query sequence_to_look_for.fasta
You can recreate any of the options available via the online interface with the right set of command line options. A full listing will be shown by running blastn -h or blastn -help.
Without any additional options, the two examples above output directly to the command line. You can direct the output to a file with the -out filename option. The output is also quite extensive, and although it is human-readable, it isn’t easy to process for a computer. There are ways to modify the output with the option -outfmt n where n is a number, and perhaps the most useful is -outfmt 6, which produces a tab-separated table summarising the hits found.
# Run blastn for nice output
blastn -db /path/to/db -query sequence_to_look_for.fasta -out blastn_results.txt -outfmt 6
The output columns, in order, are:
1. qseqid query or source (e.g., gene) sequence id
2. sseqid subject or target (e.g., reference genome) sequence id
3. pident percentage of identical matches
4. length alignment length (sequence overlap)
5. mismatch number of mismatches
6. gapopen number of gap openings
7. qstart start of alignment in query
8. qend end of alignment in query
9. sstart start of alignment in subject
10. send end of alignment in subject
11. evalue expect value
12. bitscore bit score
For more information on this output format, look here
If you are searching a very large database, blastn can take a very long time to run. There are a couple of options that you can use to improve speed:
# Use a faster but less sensitive algorithm
-task megablast
# Use more compute threads if available
-num_threads 32
Exercise 4.4
Make sure you are in your homefolder before executing the commands
# Go to your homefolder
cd
Run BLAST on the command line to determine what sequence
/nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_02.fastamight be - be careful to choose the correct algorithm
# It is always good to have a quick look at the sequence you want to find out
less /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_02.fasta
# Since we want to analyse a protein sequence with a nucleotide database, we have to use tblastn
tblastn -db /nfs/teaching/databases/ncbi/ref_prok_rep_genomes -query /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_02.fasta -out tblastn_results.txt -outfmt 6 -num_threads 32
# The sequence analysis reveals that the sequence is from the Thermus thermophilus bacterium
Run BLAST on the command line to determine the similarity between mystery_sequence_02.fasta and mystery_file_03.faa
# It is always good to have a quick look at the sequences you want to investigate
less /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_02.fasta
less /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_03.faa
# Since we want to align two protein sequences, we have to use blastp
blastp -query /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_02.fasta -subject /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_03.faa -out blastp_results.txt -outfmt 6 -num_threads 32
# The sequence analysis reveals that the sequences are 50% identical, thus there is some similarity but there are also a lot of differences in encoded amino acids
How could you have answered the previous question more computationally efficient? Hint: Remember that initially you only had the files mystery_sequence_01.fasta and mystery_sequence_02.fasta
# Since we want to compare a protein sequence (mystery_sequence_02.fasta) with a nucleotide sequence (mystery_sequence_01.fasta), we have to use tblastn
tblastn -query /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_02.fasta -subject /nfs/teaching/551-0132-00L/4_Alignment/mystery_sequence_01.fasta -out tblastn_results_2.txt -outfmt 6 -num_threads 32
# The result is the same as for the previous question
Multiple sequence alignment (MSA)¶
As we will cover in later sections, there are situations in which you want to compare and align multiple sequences all at once. This is a much harder problem to solve than pairwise alignment, in fact producing a truly optimal alignment is not feasible within a reasonable computational time, and there are various approaches that can be taken depending on what is already known about the relationships between the sequences. We will look at two approaches that make few assumptions about the sequences to be aligned, and which are used by a lot of MSA software.
Progressive alignment¶
This approach builds a final MSA by combining pairwise alignments, starting with the two closest sequences and working towards the most distantly related. The problem with this method is that part of the alignment that is optimal when it is introduced early in the process might not be so good later when other sequences join the MSA.
One popular implementation of this method is MAFFT, available here. We have also made the software available on our server and will show you the basics of how to use it here. At minimum, MAFFT requires an input file with multiple sequences in fasta format and usually always outputs to the command line, so we must redirect it.
# Run MAFFT
mafft my_sequences.fasta > my_msa.fasta
The output format is by default fasta but can be set to clustal format, explained below. Other options relate to the speed or accuracy of the aligner - you can read more in the MAFFT manual if interested.
Another popular implementation of this method is Clustal, the current version of which is called Clustal Omega and is supported by the EMBL-EBI, hosted here. We have also made the software available on our server and will show you the basics of how to use it here. At minimum, Clustal Omega requires an input file containing multiple sequences, accepting both multi-fasta and existing alignment formats.
# Run Clustal Omega
clustalo -i my_sequences.fasta -o my_msa.fasta # -i means input, -o means output
The output is by default also in fasta format, but now each sequence has gaps inserted at the right points so that the nth position in each sequence is aligned. Once again, there are many command options available, many of which won’t make any sense to you at the moment, but some are immediately useful. For instance, –outfmt allows you to select a different output format - there is no dominant format for MSA, and programs that use them as input may or may not support any specific format you choose. Clustal has a format itself which is useful for browsing a multiple alignment as it includes a line of characters indicating whether or not a column in the alignment is identical or not. The width of this format can be adjusted with –wrap.
Iterative alignment¶
Iterative methods differ from progressive alignment by going back to sequences previously introduced to the MSA and realigning them. Exactly how often and how to do these realignments varies between software packages. These methods also cannot guarantee an optimal alignment, and the trade-off versus progressive methods is that the realignments obviously take additional computational time.
A popular iterative-based method is MUSCLE, available here. We have also made this software available on our server and will show you the basics of how to use it here. At minimum, MUSCLE also only requires an input fasta file containing multiple sequences - other formats are not accepted.
# Run MUSCLE
muscle -in my_sequences.fasta -out my_msa.fasta # -in means input, -out means output
The output is by default also in fasta format, and only a few other formats are supported. Beyond that, the options determine how long the algorithm will run for - more iterations may improve the alignment but will take longer, and each incremental improvement takes longer and longer to achieve.
Exercise 4.5
Make sure you are in your homefolder before executing the commands
# Go to your homefolder
cd
Perform a multiple alignment of the file
/nfs/teaching/551-0132-00L/4_Alignment/gyra.faawith each program, obtaining Clustal formatted output, and compare the results by looking at the output files
# MAFFT
ml MAFFT
mafft --clustalout /nfs/teaching/551-0132-00L/4_Alignment/gyra.faa > mafft_aln.txt
# Clustal-Omega
ml Clustal-Omega
clustalo -i /nfs/teaching/551-0132-00L/4_Alignment/gyra.faa -o clustal_aln.txt --outfmt clu
# MUSCLE
ml MUSCLE
muscle -in /nfs/teaching/551-0132-00L/4_Alignment/gyra.faa -out muscle_aln.txt -clw
# See that there are some differences between methods