Project¶
Overview¶
This project will test your learning and understanding of the first half of the course. You must complete the work successfully to complete the project and to pass the course. The deadline for submission of your solutions is Monday 17.04.2023 23:59. In class on April 21st, we will go through an example solution, after which you will have until Wednesday 26.04.2023 23:59 to correct your solutions.
You should submit your solutions on moodle: https://moodle-app2.let.ethz.ch/mod/quiz/view.php?id=892829
Most of the solutions will be similar to those of in-class problems we have been solving each week, or can be found by starting from those exercises. We do not want anyone to struggle for a long time trying to get something to work, so please use the usual channels to seek help when you are stuck or otherwise need help:
Our slack channels https://join.slack.com/t/ethzptb/shared_invite/zt-13bdvrhzk-gHOpMuHYe4eWzaYeVh0lhg
Office hours: Monday from 14:00 - 15:00
Email: Chris Field fieldc@ethz.ch, Daniel Gehrig gehrigd@ethz.ch
You can also try our offices (HCI F409-413) but there is no guarantee that someone will be around.
Part 1¶
Each of you will find a fasta file named <your ETH user name>.fasta in the directory /nfs/teaching/551-0132-00L/7_Project/1_Genome/ - this is an individual bacterial genome specific to you that you are going to investigate. If you were to isolate a bacteria of interest in an experiment and prepare genomic DNA then a typical workflow to sequence, identify and analyse it might be:
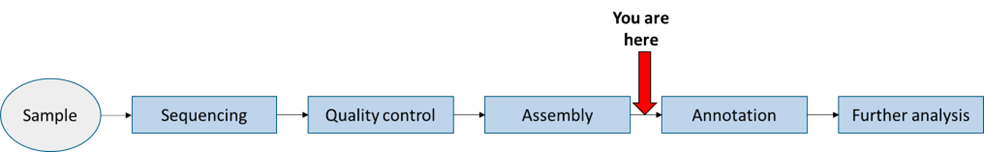
Here we are going to start at the point after assembly, where you have a genome in one or more scaffolds or contigs - these are pieces that could not be put together because either they are separate molecules (chromosome and plasmids for instance) or the assembly procedure couldn’t resolve how they should connect.
Answer the following questions about your genome:
Warning
Before reading further, copy your genome to your home folder and work from this copy - should you ever accidentally overwrite or modify your copy, you can make a new copy from the original.
Question 1
What is the total length of your genome (all scaffolds/contigs) in base pairs?
Question 2
What is the GC-content of your genome? Give your answer as a percentage to one decimal place, i.e.: 54.2%.
Question 3
Use prodigal to predict the protein-coding genes in your genome. How many genes are predicted?
Question 4
Extract the 16S sequence(s) of your genome with barrnap. Using the sequence(s), what is the taxonomic classification of your organism?
Question 5
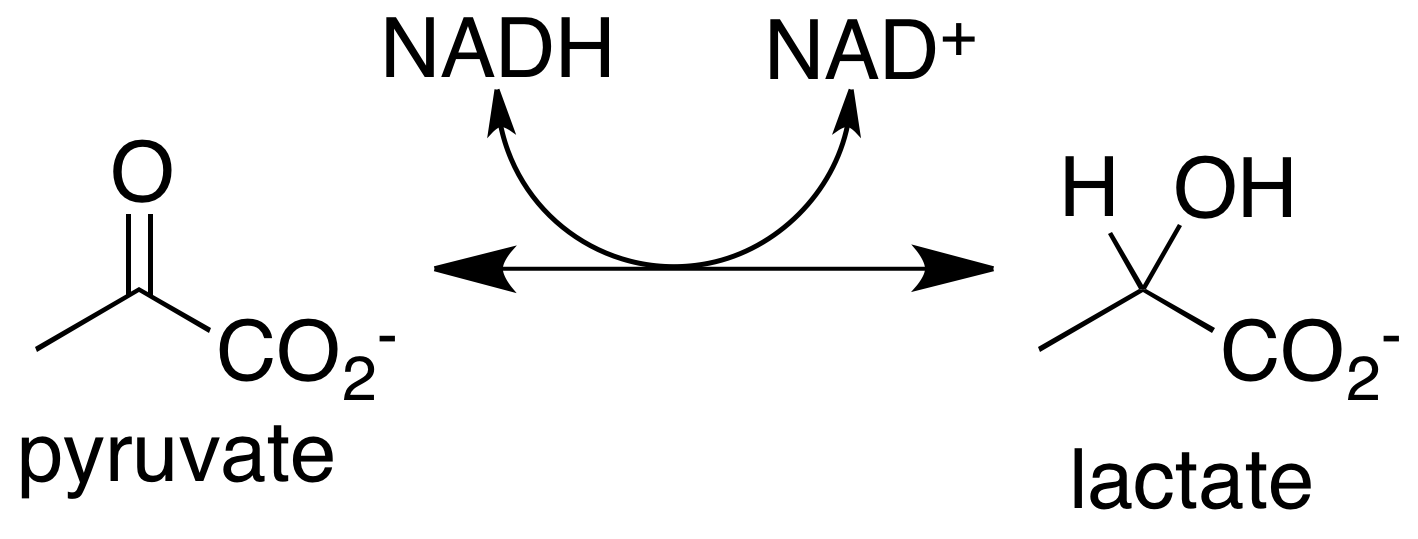
The enzyme lactate dehydrogenase or LDH, catalyses the conversion of pyruvate to lactate and back. An example from E. coli can be found here on UniProt ldhA: D-lactate dehydrogenase.
{kind=link}
What evidence can you offer to show that your organism is or is not capable of this enzymatic activity?
Part 2¶
Each of you will find a fasta file named <your ETH user name>.fna in the directory /nfs/teaching/551-0132-00L/7_Project/2_Genes - this is an individualised set of between 20 and 40 homologous gene sequences from vertebrates that you are going to investigate. Two of the sequences are homologs from the human genome, but the others could be from a variety of organisms. The data was sourced from the Ensembl database.
Answer the following questions about your gene sequences:
Warning
Before reading further, copy your gene sequences to your home folder and work from this copy - should you ever accidentally overwrite or modify your copy, you can make a new copy from the original.
Question 6
What are the names and functions of the human homologs?
Question 7
Construct a multiple alignment and phylogeny of the gene sequences. Are the two human genes nearest neighbours to one another? Which other organism(s) are nearest to the human genes?
Question 8
Consider your answer to Question 7, the number of gene homologs seen in other organisms, and the phylogeny you built. What do you deduce about the historical timing of the duplication event that created the two gene copies seen in humans?
Part 3¶
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an enveloped, positive-sense, single-stranded RNA virus that causes coronavirus disease 2019 (COVID-19). Virus particles include the RNA genetic material and structural proteins needed for invasion of host cells. Once inside the cell the infecting RNA is used to encode structural proteins that make up virus particles, nonstructural proteins that direct virus assembly, transcription, replication and host control, and accessory proteins whose function have not been determined.
ORF1ab, the largest gene, contains overlapping open reading frames that encode polyproteins PP1ab and PP1a. The polyproteins are cleaved to yield 16 nonstructural proteins, NSP1-16. Production of the longer (PP1ab) or shorter protein (PP1a) depends on a -1 ribosomal frameshifting event. The proteins, based on similarity to other coronaviruses, include the papain-like proteinase protein (NSP3), 3C-like proteinase (NSP5), RNA-dependent RNA polymerase (NSP12, RdRp), helicase (NSP13, HEL), endoRNAse (NSP15), 2’-O-Ribose-Methyltransferase (NSP16) and other nonstructural proteins. SARS-CoV-2 nonstructural proteins are responsible for viral transcription, replication, proteolytic processing, suppression of host immune responses and suppression of host gene expression.
The structural proteins of SARS-CoV-2 include the envelope protein (E), spike or surface glycoprotein (S), membrane protein (M) and the nucleocapsid protein (N). The spike glycoprotein is found on the outside of the virus particle and gives coronavirus viruses their crown-like appearance. This glycoprotein mediates attachment of the virus particle and entry into the host cell.
Source: https://www.ncbi.nlm.nih.gov/sars-cov-2/
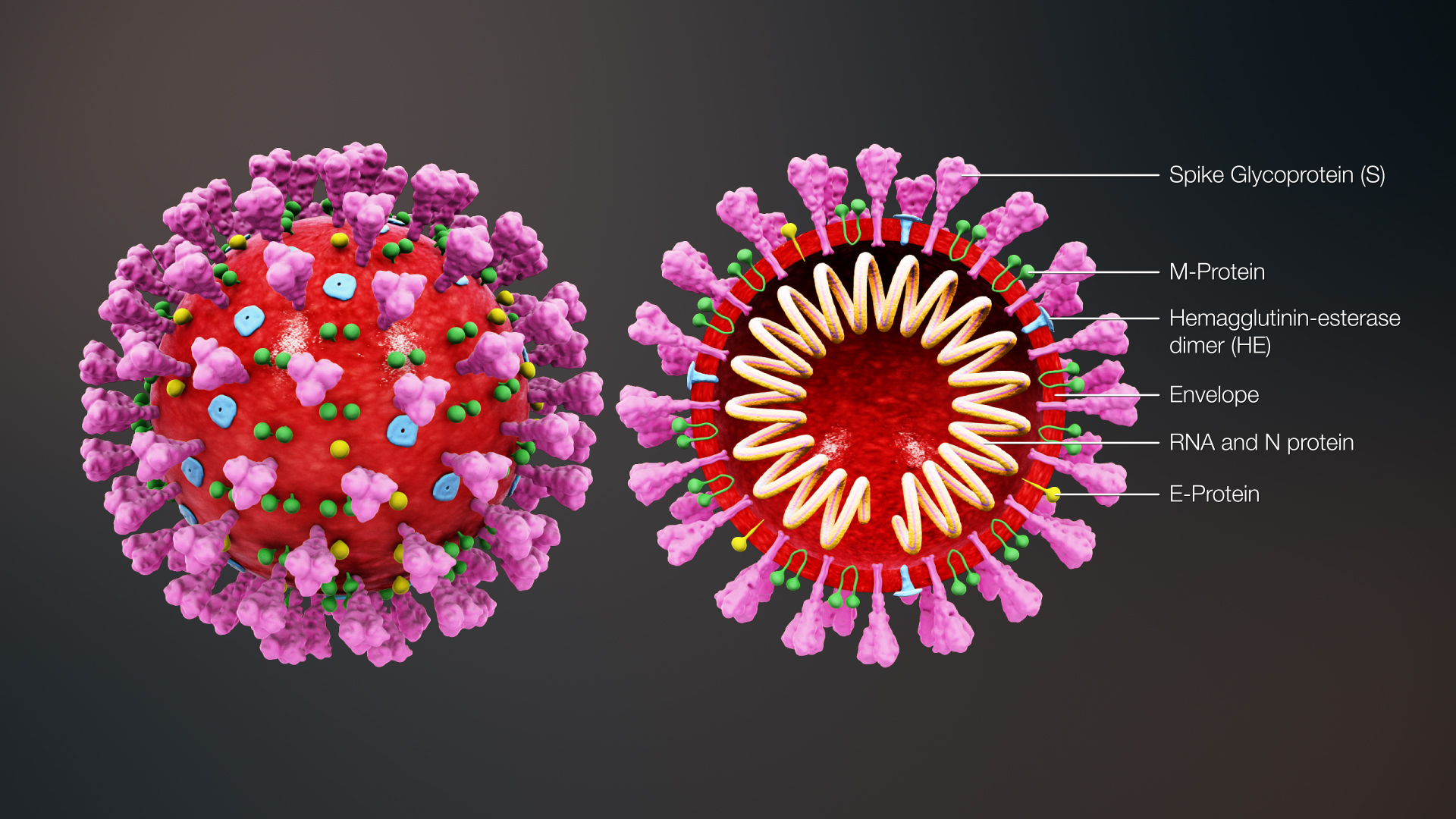
Source: https://www.prof.uzh.ch/en/news/Coronavirus-(2019-nCoV).html
You will find two fasta files called RdRp.faa and S.faa in the directory /nfs/teaching/551-0132-00L/7_Project/3_Virus - these contain collected amino acid sequences of the RNA-dependent RNA polymerase and spike glycoprotein S of sequenced Coronavirus SARS-CoV-2 samples. You will also find the reference sequence for the virus itself, SARS-CoV-2.fa, and an indexed database of reference virus sequences, RefSeq_Virus.fa.
Answer the following questions about the virus sequences:
Warning
Do not copy these files - they are significantly larger than the previous files you have worked on and it is a waste of space for you to all copy the same data. Instead you can create a symlink to the folder in your home directory as follows:
cd
ln -s /nfs/teaching/551-0132-00L/7_Project/3_Virus 3_Virus
ll # should show a line 3_Virus -> /nfs/teaching/551-0132-00L/7_Project/3_Virus
Question 9
Align the reference sequence of the virus to the provided database of virus sequences. Looking at the nearest virus in a non-human host, what is its host?
Question 10
Based on your knowledge on how the immune system works, which of the two proteins RdRp and S will have more sequence variants? Formulate a biologically meaningful hypothesis.
Question 11
How many unique sequences are in each file, RdRp.faa and S.faa?
Question 12
Consider how differences in the length of the two genes impact your results, and correct for this in your answers to Question 11. Do the final numbers support your hypothesis?