Annotation¶
General information¶
Main objective¶
In this lesson you will learn about the concept of sequence annotation. You will learn (i) how to predict genes from genome sequences, (ii) how to predict their function by homology to genes from other organisms and the databases that we use to do this, and (iii) how to automate this process.
Learning objectives¶
Learning objective 1 - You can perform gene predictions
You understand the concept of reading frames in genome sequences
You can use command line tools to predict open reading frames (ORFs) in bacterial genomes
Learning objective 2 - You can predict gene function
You understand the concept of conserved domains in proteins
You understand the homology-based function annotation of proteins
You can predict gene functions using command line tools
You learn about important DNA/protein sequence databases and their differences in quality and evidence provided
Learning objective 3 - You can automate gene function prediction
You can use command line tools to perform automated gene function prediction
Introduction to annotation¶
When we look at a DNA sequence, how do we know what it does? What features it encodes? Like most of science, we build on the work done before. If the function of a genetic element was determined experimentally in the past, and if we see that genetic element again, or something that looks like it, we might suspect that it has the same function. Then, at some point we have seen enough examples of this particular element to figure out a pattern or set of rules to predict the existence of that element in a completely new sequence. Broadly, this is known as function by homology or homology-based inference, which is based on the similarity of two genes and their functions because of their descent from a common evolutionary ancestor.
There are various different annotation tasks you might want to perform on a novel sequence, for example:
Gene prediction
Searching for a gene with a particular function
Promoter and protein binding site prediction
The prediction of various non-messenger RNAs
The prediction of CRISPR arrays
Finding signal peptide features in coding sequences
In these exercises we will show you how to predict genes in prokaryotes, how to predict gene function by homology and how to search for particular functions.
Gene prediction¶
Gene prediction in prokaryotes¶
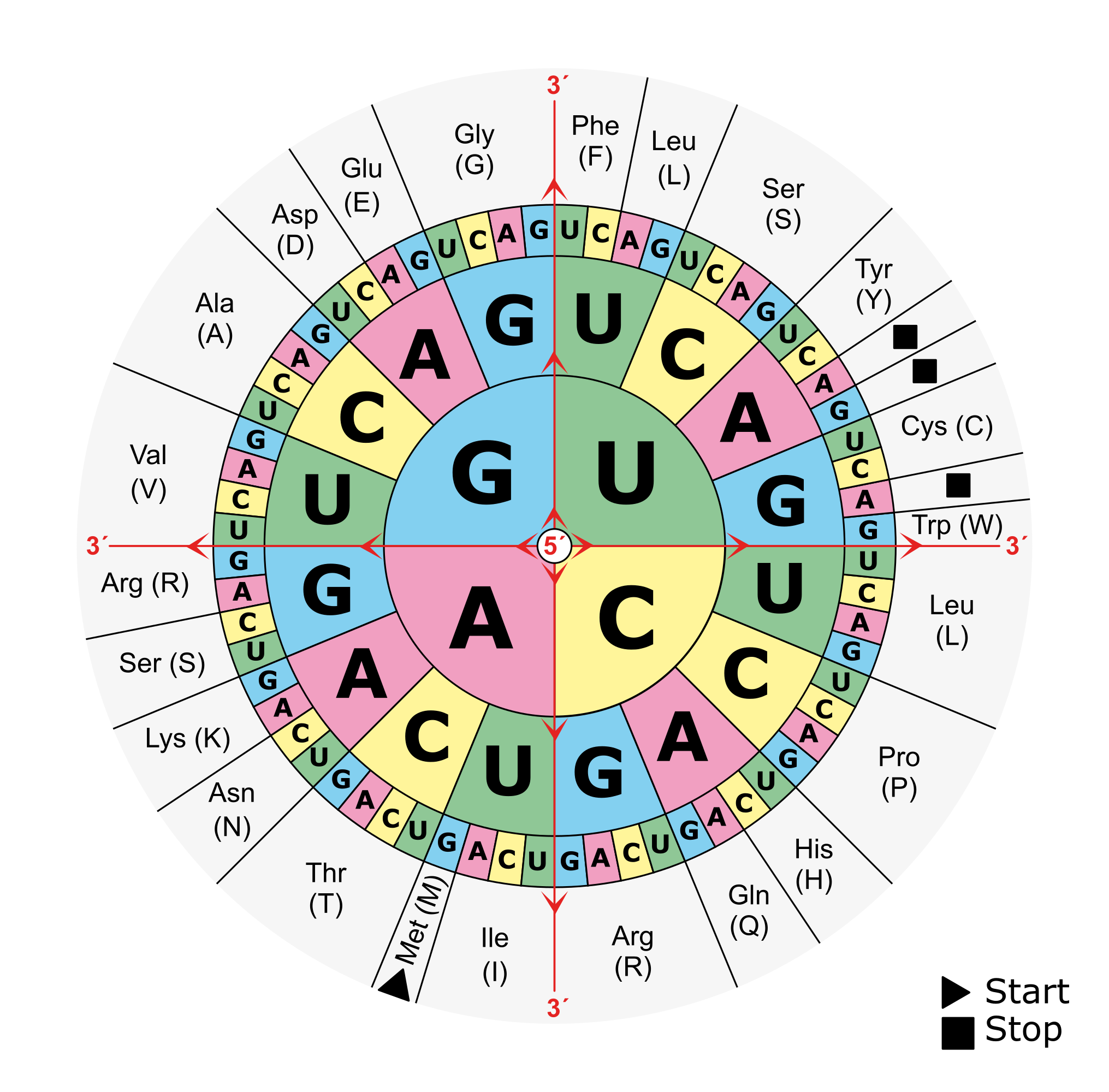
In prokaryotes, the search for genes is relatively simple: As the coding density across a prokaryotic genome is relatively high, most of the genome sequence encodes genes thus there are small intergenic regions. Genes in prokaryotes are also relatively simple in structure: they begin with a start codon, end with a stop codon and contain a ribosomal binding site (RBS) motif. When looking for a gene, there are six possible reading frames, or six possible ways to convert a nucleotide sequences into an amino acid sequence:

There are 3 frames on the top strand reading left to right, starting with the 1st, 2nd or 3rd base of the sequence in green, orange or magenta respectively. There are 3 additional frames on the complementary strand reading right to left, starting with the last, last but one or last but two base of the sequence in magenta, orange or green respectively. An open reading frame (ORF) is a region in one of these reading frames that begins at a start codon (M for Methionine) and then extends long enough that the region could be a gene before reaching a stop codon (* for any of the three possible stop codons). In this example a very small open reading frame exists, highlighted in grey. As a reminder, this is the amino acid codon table for prokaryotes, remembering that the base U in RNA corresponds to the base T in DNA:
Gene prediction algorithms look for these open reading frames and then use a model based on training data to determine which are likely genes, taking into account:
The start codon distribution
RBS motifs
Gene lengths
Overlap between genes (on the same and opposite strands)
Prodigal¶
The most popular prokaryotic gene predictor is a software package called prodigal, available here. We have also made it available on our module system (ml prodigal). Minimally, prodigal requires an input file in fasta format of at least 20kbp and will produce output directly to the command line. In our example command, instead of producing the output directly to the command line, we use the -o option to send the output to a file.
# Minimal example of running prodigal
prodigal -i my_genome.fasta -o my_genome.gbk
# Running prodigal with useful outputs
prodigal -i my_genome.fasta -o my_genome.gbk -d my_genes.fna -a my_genes.faa
In the second example we add the -d and -a options to output the nucleotide and amino acid sequences of the predicted genes to files. You can also modify the format of the main output file with -f and other options allow you to tailor the algorithm to your application, such as for a metagenome or just to train the algorithm parameters for use on another sequence.
Exercise 5.1
Run prodigal on one of the genomes you have previously worked with, either in
/nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/or one you downloaded.
# Load the module
ml prodigal
# Run the program (change the filenames for your genome)
prodigal -i my_genome.fasta -o my_genome.gbk -d my_genes.fna -a my_genes.faa
Using command line tools, count how many genes were annotated (you can use any of the output formats for this but some are easier than others).
# Count the number of predicted genes
grep -c "^>" my_genes.fna
# Use seqtk on a multi-fasta file to receive the number of genes
ml seqtk
seqtk comp my_genes.fna | wc -l
Gene prediction in eukaryotes¶
In eukaryotes, genes are more complicated features, notably with introns and exons that make the prokaryotic approach unworkable. The approach is instead to train a more sophisticated model on existing eukaryotic genes in a closely-related organism and use this model to predict genes in novel sequence. An example of software that does this is GlimmerHMM, available here. As we said at the start of the lesson Alignment within the section Comparative Sequence Analysis, it is beyond the scope of this course to look at this in more detail.
Gene function¶
Now that we have identified potential protein sequences in our genome, we should try to identify their function. To this end, we first have to understand how an amino acid sequence encoded by a gene becomes a functional protein within a cell. The translated amino acid chain (polypeptide) only represents the primary protein structure. A single polypeptide will fold into different folds due to hydrogen bonds and Van der Waals forces (most commonly alpha-helices and beta-sheets), which makes up the secondary structures. Lastly, these secondary structures will arrange themselves in the 3-dimensional space which is the tertiary structure necessary for protein functioning. Proteins with multiple subunits (f.ex. DNA gyrase with subunits A/B) additionally contain a quaternary structure, created by the interaction and arrangement of the subunits into a whole protein complex, but this is outside the scope of this course.
The overall 3D structure of a protein is typically optimized for a global minimum of an energy function which provides stability. In addition, regions within the structure which are strictly necessary for protein functioning are often conserved - similar between proteins that contain the same function. This is because they are directly involved in the functioning of that protein. Common examples for such conserved protein domains are f.ex. enzyme active sites, molecule binding sites of transportation proteins or structural components of regulatory proteins. For the protein domains to be conserved, the amino acids at specific positions need to be conserved as well. Since each amino acid sequence contains one or more of these conserved regions, gene function and protein families can be predicted based on the principle of sequence homology described in the introduction.
Gene function prediction¶
To find the function of an unknown protein, we would thus look for such conserved domains and compare amino acid sequences. There are many databases we could search to find similar sequences, but rather than just using the largest possible, we should consider the quality of evidence provided by each. For instance, RefSeq is a more carefully curated set of sequences than GenBank as a whole. Further, evidence of an actual protein sequence and function is better than evidence of only a transcript. While a transcript means that the gene is transcribed into RNA, this RNA might just have regulatory functions and not being translated into a protein necessarily. Many resources exist that aim to collect the best quality information about genes and proteins and we are going to cover a few below.
UniProt¶
A useful resource for protein sequence and function information is the database UniProt, a collaboration between the European Bioinformatics Institute (EMBL-EBI), the Swiss Institute of Bioinformatics (SIB) and the Protein Information Resource (PIR). Within this project exists Swiss-Prot, which is manually annotated and reviewed, and UniRef, with protein clusters based on sequence similarity thresholds.
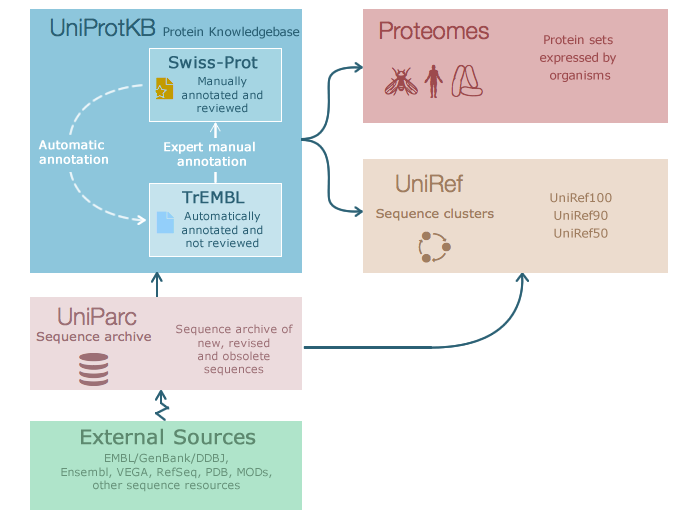
The website allows you to search by text, such as gene name or organism, or by sequence with BLAST. As a brief introduction, let’s look at the entry for a well known bacterial protein, DNA Gyrase subunit A or gyrA:
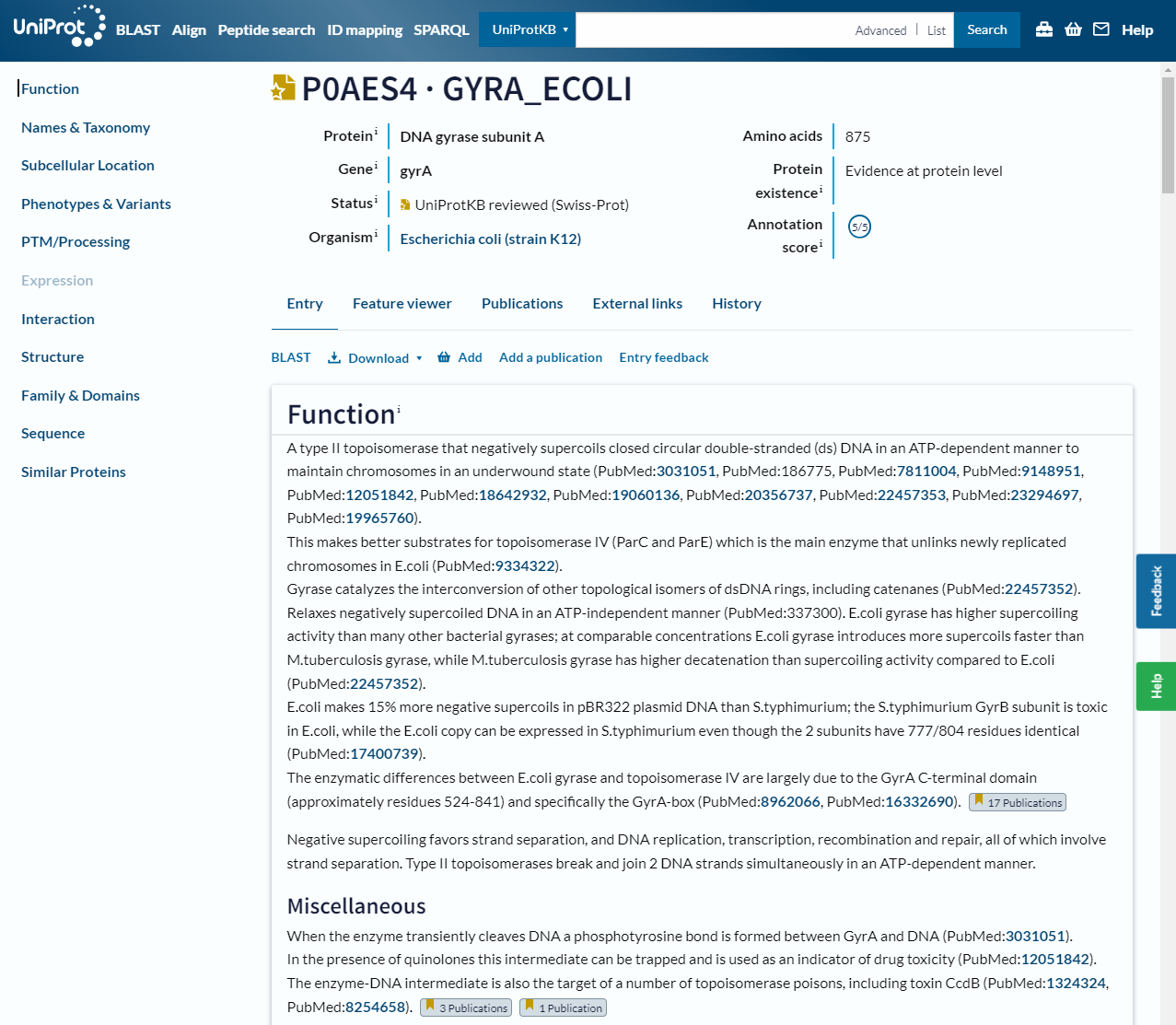
In this header you can see the gene name and organism it is from, as well as the annotation status, which gives you an idea of how much evidence there is for this particular annotation. There is also a summary of the functions of the protein with links to the scientific papers that support these statements. On the left is a table of contents for the rest of the page:
Names & Taxonomy: lists standard names, any alternative or historical names, the organism and its taxonomy
Subcellular location: shows you where in the cell the protein has been located, if evidence is available
Phenotypes & Variants: describes phenotypes and disease status associated with the protein and possible mutations of it, as well as drugs and chemicals it interacts with
PTM/Processing: lists post-translational modifications and processing of the molecule
Expression: information on the mRNA and protein levels in the cell or tissue
Interaction: describes the quaternary structure of the protein and interaction with other proteins and complexes
Structure: information on the tertiary and secondary structure of the protein that may include 3D structures from experiment or prediction
Family & Domains: information on sequence similarities with other proteins and its domains (we will discuss this more in the next section)
Sequence: the amino acid sequence of the protein as well as known variants and database listings
Similar proteins: links to other proteins in the database based on percentage identity
Cross-references: information from other databases (a summary of links in other sections)
Entry information: metadata about the protein entry itself
Miscellaneous: information that doesn’t fit into any of the other sections
As you can see, a vast amount of information is available about a single protein and UniProt does a good job of collating it all for easy access.
When it comes to annotation, we can use UniProt as a database for alignment of our unknown genes. We could also use SwissProt as a narrower but more trustworthy database, since it is manually curated. If we wanted to use broader but more hypothetical information, we could consider looking at protein domains.
InterPro¶
In the “Family & Domains” section for Gyrase A, one of the databases linked to is InterPro, also run by the EMBL-EBI and indeed based on the data in UniProt. InterPro is database used for classification of protein families, thus it groups proteins containing similar domains and important sites. InterPro is an overarching database which uses models to predict protein function based on different databases (so-called member databases). If you click on Browse and then on By Member Database you will receive an overview of existing databases used by InterPro (including the commonly used Pfam and Tigrfam databases).
InterPro is a resource that provides functional analysis of protein sequences by classifying them into families and predicting the presence of domains and important sites. To classify proteins in this way, InterPro uses predictive models, known as signatures, provided by several collaborating databases (referred to as member databases) that collectively make up the InterPro consortium. A key value of InterPro is that it combines protein signatures from these member databases into a single searchable resource, capitalising on their individual strengths to produce a powerful integrated database and diagnostic tool. InterPro further provides its entries detailed functional annotation as well as adding relevant GO terms that enable automatic annotation of millions of GO terms across the protein sequence databases.
InterPro integrates signatures from the following 13 member databases: CATH, CDD, HAMAP, MobiDB Lite, Panther, Pfam, PIRSF, PRINTS, Prosite, SFLD, SMART, SUPERFAMILY AND TIGRfams (the InterPro Consortium section gives further information about the individual databases).
The member databases use a variety of different methods to classify proteins. Each of the databases has a particular focus (e.g. protein domains defined from structure, or full length protein families with shared function).
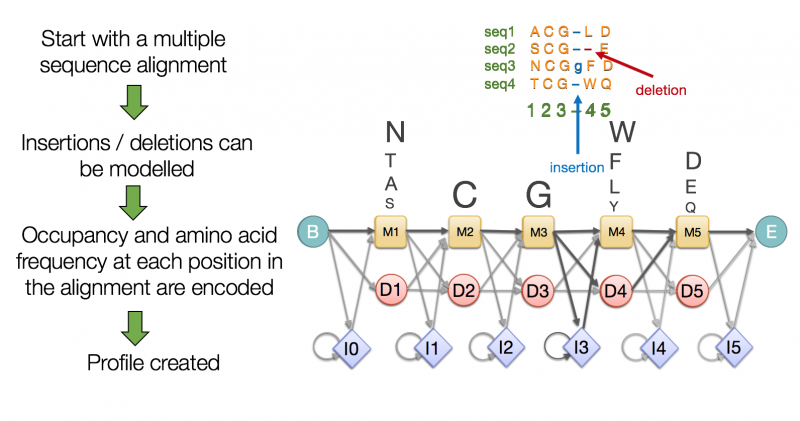
For example Pfam uses the principle of protein domains to define members of a protein family. A protein family is a group of proteins that are evolutionarily related to each other and therefore highly similar. A Pfam family consists of a small set of representative members, a multiple alignment of their sequences and a profile Hidden Markov Model (HMM) built from the multiple sequence alignment (MSA).
A Pfam HMM is a statistical model that encodes the likelihood of each amino acid at each position along with the likelihood of an insertion or deletion. You can take any sequence and score it with such an HMM to determine whether it is likely to be represented by the model or not, and therefore whether it is likely to be a member of that particular Pfam family
Let’s look at an example entry in InterPro the DNA gyrase C-terminal domain, beta-propeller which is a Pfam entry.
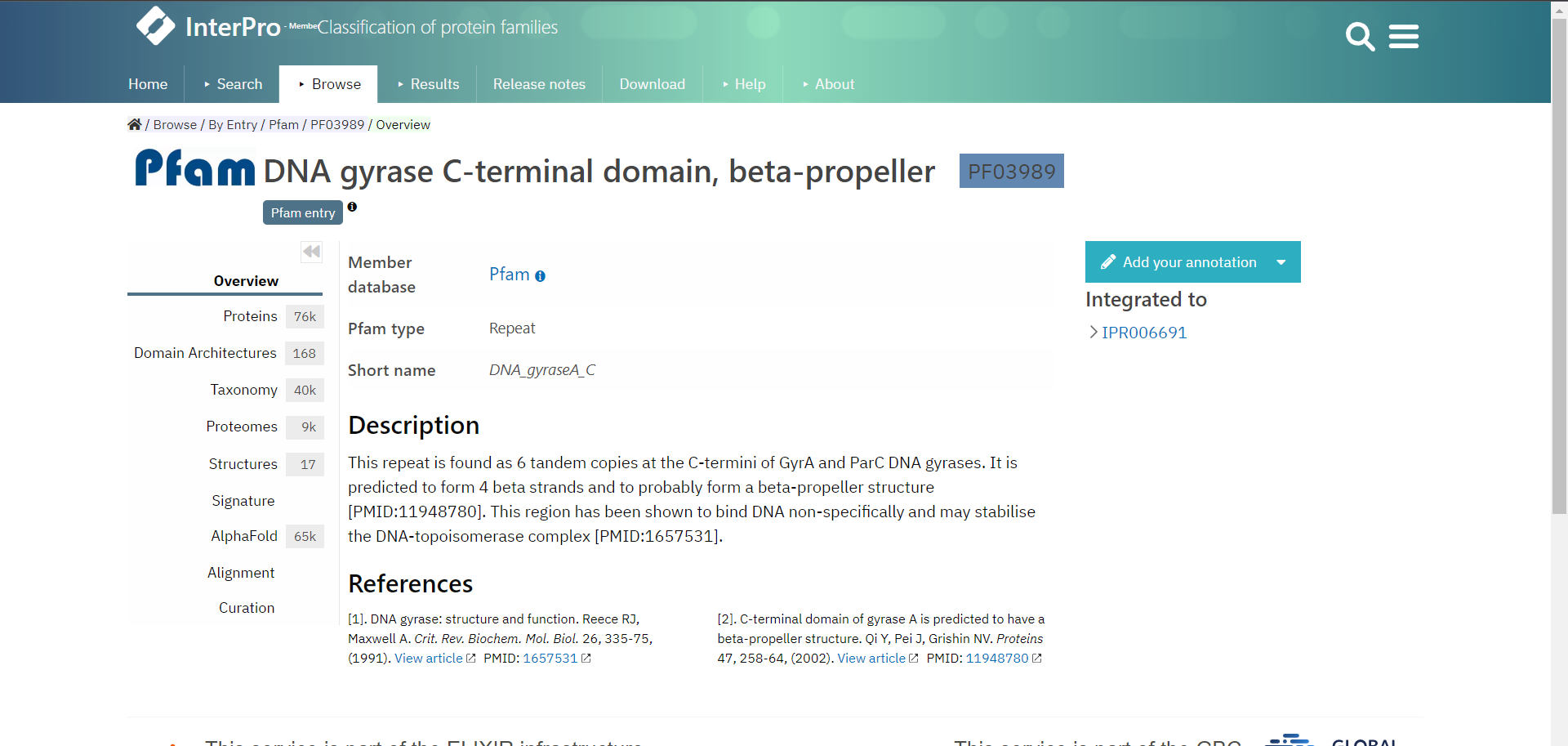
InterPro provides several useful pages of information accessible from the menu on the left, depending from which database your entry is from, the pages can vary. It is therefore the best if you explore the InterPro website yourself. However, each entry will have an Overview page which tells you from which database the entry is from and describes the entry and gives references literature to support this.
To use Pfam effectively, there is a suite of software called HMMER that can be used to both produce and search HMM profiles.
HMMER¶
HMMER is available here, and we have made it available on the module system (ml HMMER). We will quickly show you how to build and search with an HMM.
To construct an HMM from a multiple sequence alignment (MSA), you should use the program hmmbuild:
# Build an HMM - note the unusual order with the output file first
hmmbuild my_hmm.hmm my_msa.fasta
# Search sequence(s) with an HMM
hmmsearch my_hmm.hmm my_sequences.fasta
So for annotation, we could take the Pfam family database and check our sequences against it for domains, and those can inform our functional annotation.
Exercise 5.2
Annotate the sequence mystery_sequence_03.fasta found in
/nfs/teaching/551-0132-00L/5_Annotationusing UniProt and Pfam web resources
Firstly you will need to download the file to your computer using the scp command from your own machine (if you don’t remember how, go back to lesson 1 - Unix1 where we gave you an introduction to scp). Alternatively you can also open the file with less and copy/paste.
From the Uniprot website, you can perform a BLAST search (here) of the sequence against the Uniprot database. It should match a variety of XerC proteins from E. coli and Shigella.
From the InterPro website, you can perform a sequence search here but you will need to first convert the nucleotide sequence to protein sequence (for example using Python). You should find two domains in the protein, both integrases, though not exclusively phage-related as the sequence is from a bacteria.
Automated annotation¶
When you have a whole genome to annotate, you want a program to do as much as possible for you automatically. There are several pipelines available such as the NCBI Prokaryotic Genome Annotation Pipeline, PATRIC and RAST. Here we will show you how to use the whole genome annotation program Prokka, which is a pipeline that uses various feature prediction tools:
Prodigal for genes
Barrnap for ribosomal RNA
Aragorn for transfer RNA
SignalP for signal peptides
Infernal for non-coding RNA
Further it searches different databases in a specific order for protein function annotation, ranking them in order of quality:
All bacterial proteins in UniProt that have real protein or transcript evidence and are not a fragment.
All proteins from finished bacterial genomes in RefSeq for a specified genus.
A series of hidden Markov model profile databases, including Pfam and TIGRFAMs.
If no matches can be found, label as ‘hypothetical protein’.
We have made Prokka available in our module system (ml prokka). It has some recommended ways of running it, with increasing complexity:
# Load Prokka from the module system
ml prokka
# Become familiar with the command parameters by opening the help page
prokka
prokka -h
prokka --help
# Beginner
# Vanilla (but with free toppings)
prokka contigs.fa
# Moderate
# Choose the names of the output files
prokka --outdir mydir --prefix mygenome contigs.fa
# Specialist
# Have curated genomes I want to use to annotate from
prokka --proteins MG1655.gbk --outdir mutant --prefix K12_mut contigs.fa
# Expert
# It's not just for bacteria, people
prokka --kingdom Archaea --outdir mydir --genus Pyrococcus --locustag PYCC
# Wizard
# Watch and learn
prokka --outdir mydir --locustag EHEC --proteins NewToxins.faa --evalue 0.001 --gram neg --addgenes contigs.fa
Those are just examples of course, but you can see that there are many ways to customise the annotation, especially the output. Since prokka commonly creates multiple output files, you are asked to define an output directory outdir (instead of an output file) and a file name prefix for all files that will be created. The pipeline then creates files with the same prefix and standardized file extensions within that output directory. Note that prokka also uses a slightly different description of options and parameters, with two dashes followed by a whole word –outdir instead of one dash followed by one letter -o. This is a more explicit parameter description commonly used in automated pipelines which have many parameters (to avoid confusion between parameters).
You can either use the command line help page (by typing prokka) or have a look at the Prokka Github page to browse through the output files that Prokka creates.
Exercise 5.3
Run prokka on one of the genomes you have previously worked with, either in
/nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/or one you downloaded. How does the annotation differ from the official genbank record? Are there more or fewer genes?
# Load prokka
ml prokka
# Let's choose the name of the output files
prokka --outdir prokka --prefix my_genome my_genome.fasta
# We learn from this exercise that the annotation can differ in many ways, even the number of genes can be wrong. The genomes we worked with so far are very well studied and many of their annotations are based on direct observations rather than computational inference.
So far we have seen that we can use diverse databases and algorithms for genome annotation and feature prediction. Can you think of risks associated with the choices we make when annotating a genome or a set of genes?
The first and probably biggest risk of misannotation comes from the databases we use. They may contain information of different levels of evidence (i.e. there is a huge difference between experimental validation of protein function and statistical inference). It is therefore important to always be aware of the level of evidence provided by a database and not interpret the results disregarding the uncertainty of a prediction.
An additional risk regarding databases is the quality of a database and the level of manual curation that is done by the people maintaining the database. In recent years and with growing amounts of data, curation has become crucial to determine which data is useful versus useless or inaccurate data. It is therefore important to know the quality of a database to be aware of additional uncertainties.
Besides the risk of errors and functional misannotations within databases, there are plenty of differences between algorithms and how their parameters are defined. It is therefore important that we know the specific parameter settings of an algorithm of choice to be aware how it evaluates a probability of an annotated gene function.
Lastly, when we work with a specific genome of interest, we should always be aware of the information that is already known/still unknown about this organism or closely related organisms. If we work with a well-known organism, we can be more confident in the annotation since the databases probably contain more detailed and thoroughly evaluated data. If we work with a less studied organism, we should be aware that there might not be much evidence for annotation and even if we find an annotation, the risk is higher that there are wrong inferences that were not studied and corrected yet.
Annotating other features¶
Other than genes, there are techniques and software for annotating an array of other features. Many of them use similar methods as we have discussed above - build a statistical model of a feature based on example sequences and use that model to find similar features. This is typical for promoters, binding sites and other sequence motifs.
In-class problems¶
In-class Problems Week 5
Some simple features encoded by DNA can be found without complex models. Using command line tools and Python, how might you:
Search for exact matches to a restriction enzyme recognition site? For instance, find the recognition site for EcoR1 online and then search for sites in the bacterial genomes in
/nfs/teaching/551-0132-00L/1_Unix1/genomes.Search for primer binding sites, allowing for one base mismatch? For instance, find the sequences for the 27F and 1492R primers that amplify the 16S rRNA and look for binding sites in the bacterial genomes in
/nfs/teaching/551-0132-00L/1_Unix1/genomes.
Imagine you had identified a new prokaryote and were interested in classifying its metabolic capabilities. With the introduced databases and methods in mind, consider the following questions:
Is your organism capable of breaking down glucose into pyruvate by glycolysis?
Does your organism encode for the enzymes required for lactose fermentation?