Sequence data¶
General information¶
Topic overview¶
In this lesson you will be introduced to the sequence data formats fastq and fasta. You will learn how features such as genes are annotated in genbank and gff formats. Sequence and feature data is available via any number of online databases, and you will explore NCBI resources.
Learning objectives¶
Learning objective 1 - You can interpret files containing biological sequence information in standard formats
You familiarise yourself with the different file formats (fasta, fastq, GenBank, GFF)
You extract information on the command line from the files
You use BioPython to manipulate the files
Learning objective 2 - You are able to navigate and search important public databases of biological information
You inspect major sequence databases (NCBI, ENA, DDBJ)
You experiment with the Entrez search system of the NCBI
You download files from databases with the command line
Sequence data¶
DNA and protein sequences are fundamental to our understanding of biology and with the advent of high-throughput sequencing technologies, we generate ever more sequencing data at an ever increasing rate. Sequence data is nowadays involved in almost every biological project in one form or another and it is consequently very likely that you will encounter and work with sequence data if you stay in biology (designing a primer, working with CRISPR or checking the genotype of a mouse; all these tasks use sequence data even though they are not directly linked to bioinformatics). Therefore, it is crucial to understand this type of data.
At the core of our ability to use this data are two standard file formats:
FASTQ - this is data from DNA sequencing that includes a quality score for each base
FASTA - this is processed data, and can be DNA or protein sequence
FASTQ format¶
A fastq file may contain multiple sequences in fastq format. This is a text-based format where each entry consists of 4 lines:
The sequence identifier, probably automatically generated by the sequencing device; the line always begins with @
The raw sequence, consisting of A, C, G, T and perhaps N (standing for bases that cannot be clearly assigned)
These days unused, but in theory a repeat of line 1; the line always begins with +
The quality score for each base in the raw sequence
@071112_SLXA-EAS1_s_7:5:1:817:345
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
Quality scores¶
Quality scores in the standard Phred format range between 0 and 93, although sequencing read data is rarely higher than 60. The scores are encoded such that there is one character per base in the 2nd line of the fastq entry, using the ASCII encoding table (a computing standard). For instance, the letter A has a decimal equivalent of 65. Sanger format offsets the quality score by 33, so this represents a quality score of 32 (65-33).
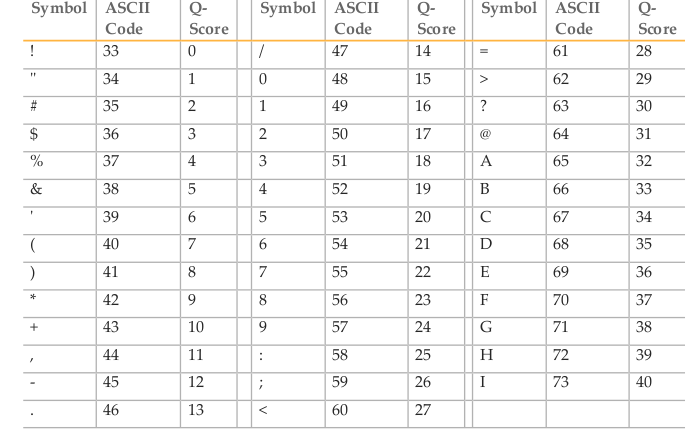
The Phred quality score (Q) is logarithmically (base 10) related to the error probability (E) and can therefore be interpreted as an estimate of error (E) or as an estimate of accuracy (A).
This formula gives us the following benchmarks for accuracy:
Phred Quality Score |
Error |
Accuracy |
|
|---|---|---|---|
10 |
10% |
90% |
|
20 |
1% |
99% |
|
30 |
0.1% |
99.9% |
|
40 |
0.01% |
99.99% |
|
FASTA format¶
A fasta file may also contain multiple sequences in fasta format (sometimes known as multi-fasta). The sequences will be either DNA or protein sequences, but never mixed in the same file. The fasta format is text-based, where each sequence entry consists of 2 elements:
A header line beginning with the > character and some information about the sequence such as ID, species name, gene name or position
One or more lines of sequence data. When containing multiple lines, each line is usually 70 or 80 characters long, and this is done only for readability.
# FASTA format of nucleotide sequence
>Mus_musculus_tRNA-Ala-AGC-1-1 (chr13.trna34-AlaAGC)
GGGGGTGTAGCTCAGTGGTAGAGCGCGTGCTTAGCATGCACGAGGcCCTGGGTTCGATCC
CCAGCACCTCCA
# FASTA format of protein sequence
>gi|5524211|gb|AAD44166.1| cytochrome b [Elephas maximus maximus]
LCLYTHIGRNIYYGSYLYSETWNTGIMLLLITMATAFMGYVLPWGQMSFWGATVITNLFSAIPYIGTNLV
EWIWGGFSVDKATLNRFFAFHFILPFTMVALAGVHLTFLHETGSNNPLGLTSDSDKIPFHPYYTIKDFLG
LLILILLLLLLALLSPDMLGDPDNHMPADPLNTPLHIKPEWYFLFAYAILRSVPNKLGGVLALFLSIVIL
GLMPFLHTSKHRSMMLRPLSQALFWTLTMDLLTLTWIGSQPVEYPYTIIGQMASILYFSIILAFLPIAGX
IENY
Exercise 3.1
/nfs/teaching/551-0132-00L/3_Sequence/ contains example files example_reads_R1.fastq, example_reads_R2.fastq and example_sequences.fasta.How might you count the number of entries (sequences) in a multi-fasta file using command line tools? HINT: Think carefully about the ways your method might go wrong, consider using a regular expression.
# Enter directory
cd /nfs/teaching/551-0132-00L/3_Sequence/
# Count fasta records
grep -c "^>" example_sequences.fasta # count lines starting with ">" within fasta file
# Description of command
# grep "", searches for regular expressions within quotation marks ""
# -c (option), prints the total count of occurences of the searched pattern instead of selected lines
# ^, means 'start of line'
# ">", searches for lines starting with >
# example_sequences.fasta (argument), the file that the command should be executed on
How about for a fastq file? HINT: Think carefully about the ways your method might go wrong, consider using a regular expression.
# Count fastq records
grep -c "^+$" example_reads_R1.fastq # count lines that contain only one "+" from start to end of line within fastq file (3rd line)
(expr $(wc -l example_reads_R1.fastq | cut -d " " -f 1) / 4) # this method is a bit complicated
# Description of command
# grep -c "" # count occurence of pattern within quotation marks ""
# ^ # means 'start of line'
# $ # means 'end of line'
# Description of more complicated method
# wc -l example_reads_R1.fastq # prints total number of lines in file and filename (separated by whitespace, because can be used on multiple files)
# \| # pipe is used as connector between two commands so that second command can be executed on output of first command directly
# \| cut -d " " -f 1 # uses output of previous command, separates output by whitespace and prints only first field/column (so only number of lines, without filename)
# expr $( ) # evaluates expression within brackets and give result
# (expr $() /4) # divides result by 4 because every fastq entry contains 4 lines
In the example fastq entry shown above
example_reads_R1.fastq, calculate the Phred quality scores for the final 4 bases.
# Quality score calculation
# Final 4 bases have the symbols 4, %, ), @
# In ASCII code that's: 52, 37, 41, 64
# In Phred (ASCII-33) that's: 19, 4, 8, 31
Feature data¶
As well as the sequence of biological molecules, it is useful to keep a record of identified features that exist as part of the molecule, whether they have been experimentally or computationally determined. For instance, the open reading frames of the genes in a bacterial genome. Here we cover two principle formats that are used to store feature information.
Genbank flat file format¶
The GenBank flat-file is designed to contain a large and varied amount of information on DNA or RNA sequences. We are not going to cover all of the possible features of the format here, but the NCBI provides a sample record with a detailed description of each component at the following link: SAMPLE RECORD.
- Locus:
Locus name: Originally had a set format but now just has to be a unique name for the sequence record.
Sequence length: Number of base pairs or amino acids.
Molecule type: For instance DNA or mRNA, from a limited set of valid types.
GenBank division: A three letter designation such as PRI (primate), PLN (plant) or BAC (bacteria), from a limited set of valid designations.
Modification date: When the record was last updated.
Definition: A brief description of the sequence such as source organism, gene name/protein name, or some description of the sequence’s function.
Accession: A unique identifier for each record that never changes (but the record may become redundant). There are several different formats for historical reasons, for example: U12345, AF123456.
Source: Organism name and sometimes molecule type. Under Organism you can find the formal scientific name for the source organism and its lineage.
- Features: Information about genes, gene products and biologically relevant regions. Each feature has:
Type: The type of feature such as gene or CDS, from a limited set of valid types.
Position: The start and end of the feature, possibly multiple start/ends for eukaryotic genes for instance.
Qualifiers: Various additional pieces of information such as /product (product name) or /translation (amino acid sequence), from a limited set of valid qualifiers.
Origin: Optionally, the full sequence of record may be included here.
GFF format¶
The GFF (General Feature Format) format is used in bioinformatics to describe genes and other features of DNA, RNA and protein sequences. The gff file consists of one line per feature, each containing 9 columns of data (fields) separated by tab characters. The 9 fields have the following properties (in the shown order).
Sequence name: name of the sequence where the feature is located.
Source: name of the program that generated this feature or the data source for the feature.
Feature: feature type name such as gene or exon, etc.
Start: start position of the feature, sequence numbering starts at 1.
End: end position of the feature.
Score: a numeric value indicating the confidence of the source in the annotated feature; “.” indicates a null value.
Strand: defined as + (forward), - (reverse) or . (undetermined).
Frame: indicates the first codon position for CDS features, 0, 1 or 2 for first, second or third position; otherwise “.”.
Attributes: additional information about the feature, separated by ;.
# Excerpt from an example GFF file for E. coli MG1655
NZ_AYEK01000001.1 RefSeq region 1 4638920 . + . ID=id0;Dbxref=taxon:511145;gbkey=Src;genome=genomic;mol_type=genomic DNA;strain=K-12;substrain=MG1655
NZ_AYEK01000001.1 RefSeq gene 190 255 . + . ID=gene0;Name=P370_RS01000000122250;gbkey=Gene;gene_biotype=protein_coding;locus_tag=P370_RS01000000122250
NZ_AYEK01000001.1 Protein Homology CDS 190 255 . + 0 ID=cds0;Parent=gene0;Dbxref=Genbank:WP_001386572.1;Name=WP_001386572.1;gbkey=CDS;inference=COORDINATES: similar to AA sequence:RefSeq:NP_414542.1;product=thr operon leader peptide;protein_id=WP_001386572.1;transl_table=11
NZ_AYEK01000001.1 RefSeq gene 337 2799 . + . ID=gene1;Name=P370_RS0100015;gbkey=Gene;gene_biotype=protein_coding;locus_tag=P370_RS0100015
NZ_AYEK01000001.1 Protein Homology CDS 337 2799 . + 0 ID=cds1;Parent=gene1;Dbxref=Genbank:WP_001264707.1;Name=WP_001264707.1;gbkey=CDS;inference=COORDINATES: similar to AA sequence:RefSeq:WP_005124053.1;product=bifunctional aspartokinase I/homoserine dehydrogenase I;protein_id=WP_001264707.1;transl_table=11
Exercise 3.2
/nfs/teaching/551-0132-00L/3_Sequence contains example files example_features.gff and example_features.gbk.How might you use command line tools to count the number of different features in a Genbank format file (.gbk)? Look at a genbank file for feature examples and design a regular expression to capture the feature names you see.
# For the example Genbank file we need to find the feature types with a clever regular expression (that looks for the correct spacer followed by letters and another space)
grep -oP '^\s{5}[a-zA-Z]+\s' example_features.gbk | tr -d ' ' | sort | uniq -c
# Description of command
# grep -oP # grep searches for pattern, -o option only outputs parts of lines that matches pattern (default is full line that matches pattern), -P allows for a more complicated Perl regex pattern
# '^\s{5}[a-zA-Z]+\s' # means line is starting with 5 spaces, followed by 1 or more letters (small or capital) and followed by one space
# tr -d ' ' # -d option deletes a pattern, here all whitespaces ' '
# sort # sorts matches alphabetically
# uniq -c # prints total count of each pattern (here feature types)
# 4302 CDS
# 4609 gene
# 79 ncRNA
# 22 rRNA
# 1 source
# 86 tRNA
How about for a GFF file (.gff)?
# For GFF format things are a bit easier as after the comment lines it is just a tab-delimited table
cut -s -f 3 example_features.gff | sort | uniq -c
# Description of command
# cut -s -f 3 # -s option leaves out the comment lines because they do not have delimiters, -f selects 3rd field/column
# sort # sorts output alphabetically
# uniq -c # prints total count of each output (here feature types)
# 4324 CDS
# 187 exon
# 4464 gene
# 50 mobile_genetic_element
# 79 ncRNA
# 1 origin_of_replication
# 164 pseudogene
# 1 region
# 22 rRNA
# 48 sequence_feature
# 86 tRNA
# See how the information in the two files is slightly different due to different format specifications
Working in BioPython¶
BioPython is an extensive package that contains freely available tools for biological computation written in Python by an international team of developers. It provides containers and functions for working with these file formats and more. To use BioPython, you should be familiar with basic programming in Python. Here you will learn how to use the package to read and manipulate sequence records.
For this part you have to work with Python. If you have forgotten how to switch to Python on the Jupyter-Hub you can find a description here.
If you have never worked with Juypter-Notebooks before, we recommend you to have a look at the Notebook Jupyter_Introduction.ipynb, which gives you a short introduction to Juypter-notebooks.
There is also a notebook (with solutions) that can help you to refresh your python skills: exercise00_introduction.ipynb for the exercise and solution_exercise00_introduction.ipynb for the solution. These notebooks were created by Andre Kahles, whom you may get to know in the Concept Course in Bioinformatics.
To do these notebooks you have to copy them from the /nfs/nas22/fs2202/biol_micro_teaching/551-0132-00L/3_Sequence folder to your home folder (in the terminal while logged into Cousteau). Therefore, open a terminal and ssh into Cousteau and run the following commands:
# Make sure you are in a terminal and run the following commands
cp /nfs/nas22/fs2202/biol_micro_teaching/551-0132-00L/3_Sequence/Jupyter_Introduction.ipynb ~/
cp /nfs/nas22/fs2202/biol_micro_teaching/551-0132-00L/3_Sequence/*00_introduction.ipynb ~/
Back in the Jupyter-Hub the files should appear in your home folder (orange square). You can work with them by simply clicking onto the files (in the Juypter-Hub)
For the upcoming exercises you have to open a new Juypter-notebook and work in there
Loading the package¶
There are two main components you are likely going to use, the Seq object from the Seq module (confusing, yes) and the SeqIO object (see next section).
The Seq object combines a Python string with biological methods relevant to sequence-specific information. Thus by saving a string of letters as a Seq object (e.g. a DNA sequence), you can retrieve the complementary strand, the transcribed RNA sequence or the translated protein sequence. When using these methods, keep in mind that Biopython does not know whether a sequence object “AGCTAGGT” you created is a DNA sequence or a protein sequence rich in alanines, cysteines, guanines and threonines.
You load the packages for these objects with standard python syntax:
from Bio.Seq import Seq
from Bio import SeqIO
Seq objects¶
To declare a new Seq object is straightforward:
my_seq = Seq("AGCTTTTCATTCTGACTG")
In many ways, Seq objects behave like strings, with find and count methods:
# Find the first position of a particular subsequence
my_seq.find("ACT")
my_seq.find("AAA") # returns -1 if not found
# Count the number of occurrences of a particular subsequence
my_seq.count("A")
my_seq.count("TT") # only non-overlapping sequences are counted
They also have useful, sequence-specific methods:
# Complement
my_seq.complement() # complementary sequence
# Reverse complement
my_seq.reverse_complement() # complementary sequence with reversed order of bases
# Transcription and reverse transcription
my_rna = my_seq.transcribe() # transcription from DNA to RNA
my_dna = my_rna.back_transcribe() # reverse transcription from RNA to DNA
# Translation works on both DNA and RNA
my_rna.translate() # translation from RNA to protein
my_dna.translate() # like transcription from DNA to RNA and translation from RNA to protein
Sequences can also be concatenated (merged) like strings, thus multiple sequences can be added after each other to create one large sequence. Furthermore, sequences can be sliced (cut into pieces) like strings. By choosing an index range of bases you can extract shorter subsequences from one large sequence. For slicing sequences, remember that Python uses 0-based indexing, so whenever you want to generate a subsequence, your index count will start from 0 thus the first letter in a sequence will be my_seq[0] (not my_seq[1]).
# Add some made up sequence
my_newseq = Seq("ATG") + my_seq
# Get the first 10bp
my_subseq = my_seq[0:10]
# Get the last 10bp
my_subseq = my_seq[-10:]
More information about Seq objects can be found here.
Reading files with SeqIO¶
SeqIO provides a simple interface to read and output sequence file formats (also multi-fasta files) in a uniform manner. SeqIO provides a function parse() that allows you to read a multi-fasta file by specifying the file name and file format. Thus parse() allows you to break down sequence files into their components by creating an iterator of SeqRecord objects (which you learn more about below).
Instead of using the file name, you can use a handle to specify which file you read. A handle is typically a file opened for reading, but it can also be an output of a previous command or data from the internet. The advantage of using a handle is guaranteeing that the file is closed correctly after reading.
# As an iterator
records = SeqIO.parse("myfile.fasta", "fasta")
# Using a handle
with open("myfile.fasta") as handle:
for record in SeqIO.parse(handle, "fasta")
<do things>
Records read in by SeqIO are SeqRecord objects, which contain a seq variable that is a Seq object and additional information such as the record ID and description. Many of the methods for Seq objects work identically for SeqRecords. More information about Seq records can be found here.
Sometimes you don’t want to work through the records in file order, in which case you can use list() to convert the iterator to a python list, but be careful with very large files as this will put every record into memory at the same time. You can also convert the iterator to a dictionary with record IDs as keys using a provided function.
# As a list object
records = list(SeqIO.parse("myfile.fasta", "fasta"))
# As a dictionary
records = SeqIO.to_dict(SeqIO.parse("myfile.fasta", "fasta"))
Note that the SeqIO.parse examples above specify the file format as “fasta”. Many other formats are supported, but the correct format must be explicitly given as an argument, for instance fastq is “fastq” and GenBank is “genbank” or “gb”. Sadly, GFF format is not yet supported and requires an additional package or parsing it yourself. The full list of formats is available here.
Accessing feature information¶
If you import a GenBank file with SeqIO, the Seq object will also contain information about the record’s features, stored as SeqFeature objects.
# Import a genbank file and inspect its features
records = list(SeqIO.parse("myfile.gbk", "gb"))
record = records[0]
# List of features
record.features
# Inspect a feature
print(record.features[1])
record.features[1].location
record.features[1].qualifiers
# Extract the sequence for the feature from the parent record
feature_seq = record.features[1].extract(record)
As features are a list, you can of course sort them using list comprehension by type, position, or similar. Note that when you slice a sequence to create a subsequence, only features that are contained completely within the subsequence are kept by it.
Writing files with SeqIO¶
SeqIO can also be used to output records to file, in the supported format of your choice. Obviously if you convert file format you might lose information, for instance fastq to fasta, or genbank to fasta. Again, the file can be written using a handle if desired.
# Write to fasta
SeqIO.write(records, "myrecords.fasta", "fasta")
# Write to fasta with a handle
with open("myrecords.fasta", w) as handle:
SeqIO.write(records, handle, "fasta")
Converting file formats¶
If you use SeqIO to read in a file in one format, you can convert it by writing to another format. There are some things to note when doing this however:
If you output to a format that does not support features, such as fasta, then you lose that information
If you extract a feature sequence or slice a sequence, the new SeqRecord inherits the additional properties such as ID and description of the parent sequence
If you translate a SeqRecord from nucleotide to amino acid sequence, the additional record information such as ID and description are lost and replaced with awkward ‘<unknown x>’ strings
Exercise 3.3
Using SeqIO, read in the GenBank file located at
/nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.gbff
# Read in the file
from Bio import SeqIO
records = list(SeqIO.parse("/nfs/teaching/551-0132-00L/1_Unix1/genomes/bacteria/escherichia/GCF_000005845.2_ASM584v2/GCF_000005845.2_ASM584v2_genomic.gbff", 'gb'))
record = records[0]
What is the GC content (the percentage of bases that are G or C) of the genome?
# Calculate GC content
gc = (record.seq.count('G') + record.seq.count('C')) / len(record)
gc # GC content is 50.8%
How many genes are there in the genome?
# Count genes
genes = [feature for feature in record.features if feature.type=='gene']
len(genes) # 4609 genes
Pick any gene and write the sequence out to a new fasta file
# Output a gene
my_gene = genes[0]
my_gene_seqrec = my_gene.extract(record) # Note the difference between a feature and a sequence record containing features
my_gene_seqrec.id = my_gene.qualifiers['gene'][0] # Change the ID of the new sequence record
my_gene_seqrec.description = 'extracted from ' + my_gene_seqrec.description # Change the description of the new record
SeqIO.write(my_gene_seqrec, 'my_gene.fna', 'fasta')
For the same gene, write the translated amino acid sequence out to another fasta file
# Output a translation
my_gene_trans = my_gene_seqrec.translate()
my_gene_trans # See that the metadata is messed up
my_gene_trans.id = my_gene_seqrec.id # Copy the metadata from the original sequence record
my_gene_trans.description = my_gene_seqrec.description # Ditto
SeqIO.write(my_gene_trans, 'my_gene.faa', 'fasta')
Note: More thorough explanations and additional material about Biopython can be found here. (it is worth a look)
Sequence databases¶
In your future work, you might want to reference the genome or some of the genes of the organism you are working with, or those of species it is related to. If you generate sequence data, you might want to identify or annotate those sequences using bioinformatic methods that rely on an evidence base of existing public sequence data. It is therefore important that you are aware of the available databases that you might browse or search for such information.
There are three primary sequence databases that essentially contain the same data, exchanged daily between them.
There are additionally a vast array of secondary databases, often specialising in particular types of sequence or individual organisms. We will discuss some of them in future parts of the course.
NCBI¶
The National Center for Biotechnology Information (NCBI) hosts a series of databases and tools that are considered essential for modern biology.
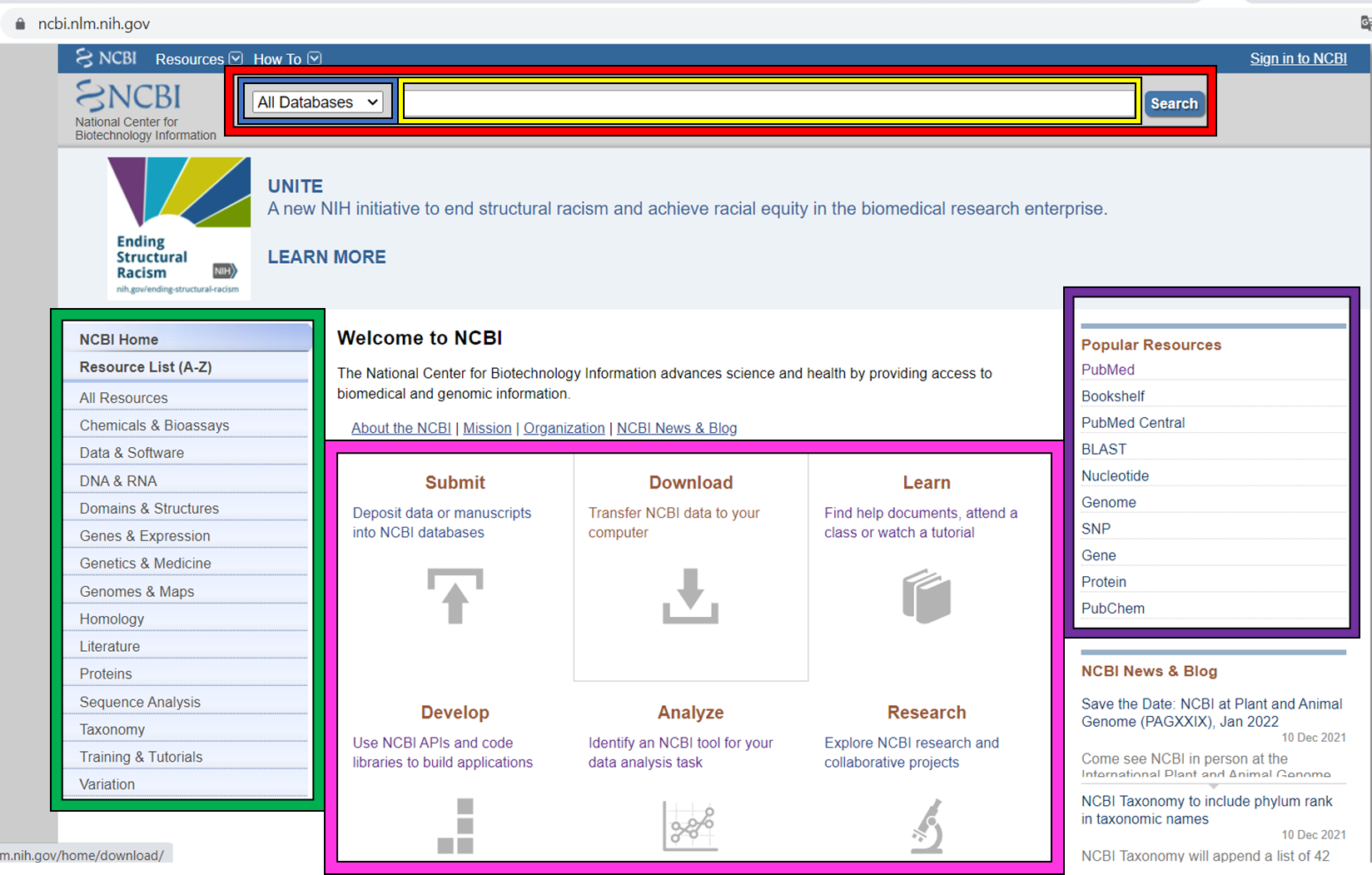
The NCBI homepage (below) is a bit overwhelming. At the top you have the search bar (red frame). You can either search (yellow frame) in all databases or you can select a specific database out of the 39 available databases (blue frame).
In the bottom half of the page you have some popular resources on the right side (purple frame) and on the left hand side (green frame) you find a variety of sub areas. In the middle (pink frame) other common features are linked.
In the following section we will describe certain parts of the NCBI to help you find what you are looking for.
GenBank¶
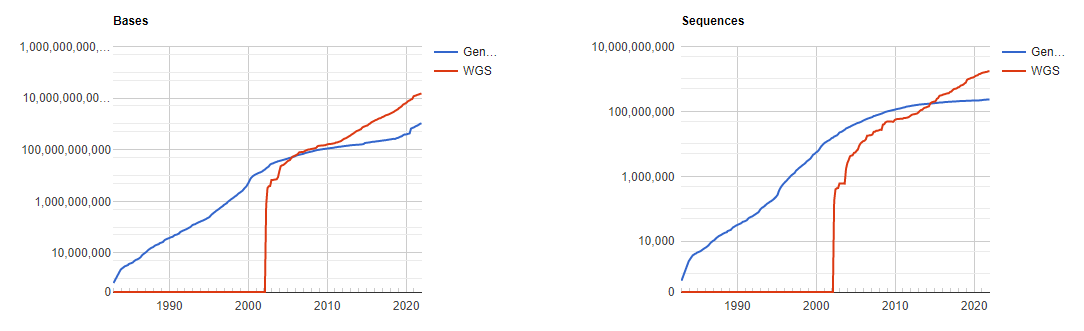
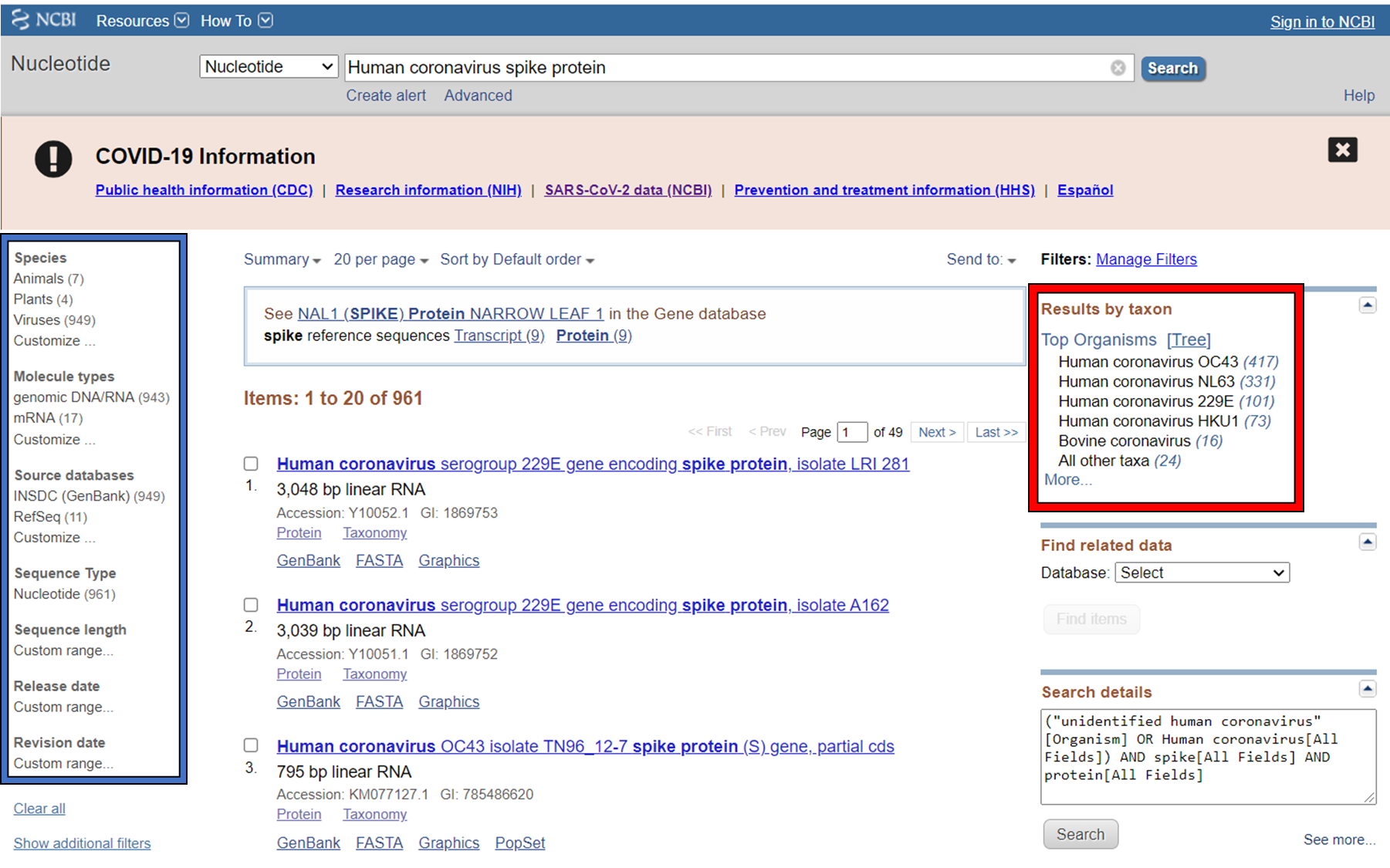
GenBank is an annotated collection of all publically available DNA sequences. This includes genomes, individual genes or feature sequences, transcripts and more. Sequences shorter than 200bp, that aren’t based on a real molecule (for instance a consensus sequence) or that are not known in nucleotide space (for instance a directly sequenced protein), primers, and mixed DNA/mRNA sequences are not accepted. Additional to GenBank is the WGS (whole genome shotgun) database, which contains sequencing projects that are currently the most common form of high-throughput sequencing, but are not yet assembled, finished or annotatable. The graphs below show how the databases have grown over time in number of entries and total base pairs.
GenBank is searchable by selecting the ‘Nucleotide’ database on the NCBI homepage. It can also be searched by alignment, which will be covered in the next lecture. When you search, you are shown the results as seen below. These can be further filtered by convenient links on the left side of the page (blue frame), or by organism on the right side of the page (red frame).
RefSeq¶
The Reference Sequence database aims to be a comprehensive, well-annotated, non-redundant set of sequences - effectively a curated subset of GenBank to represent the best quality information available for use in biological research. For instance, RefSeq contains 66,541 bacterial entries as of release 2007. If you are looking for a high quality and trustworthy sequence for your work, RefSeq is a good place to start.
RefSeq is not searchable from the NCBI frontpage. Instead, you can search GenBank by selecting the ‘Nucleotide’ database and then use the appropriate filter.
Genome¶
The genome database is another subset of GenBank that includes genomes, chromosomes and assemblies. It aims to assign taxonomy to each entry and give an assessment of completeness. It can be searched directly from the NCBI frontpage by selecting ‘Genome’.
Taxonomy¶
The taxonomy database is a curated classification of the organisms in GenBank, by which we mean their locations on the tree of life. There are alternative taxonomies available, such as the GTDB, as phylogenetic methods differ. Taxonomy is continually under revision, and often submissions are unintentionally misassigned, so be wary when working with less well researched organisms or environments.
Taxonomy can be searched directly from the NCBI frontpage by selecting ‘Taxonomy’.
Searching the NCBI¶
The NCBI’s primary text search and retrieval system, Entrez, comprises 39 molecular and literature databases and is usually accessed via the search bar (Figure 1 red frame, nearly all search boxes on NCBI access the Entrez system).
Since Entrez searches in a vast amount of databases and the search input can be almost anything (single words, short phrases, sentences, database identifiers, gene symbols, names, etc.) even simple searches can lead to an overwhelming amount of results. Therefore it is useful to know some tricks which make searching more efficient.
Boolean Operators: You should be familiar with Boolean Operators from Statistics. They can be used in Entrez to make your search more specific:
AND: Finds documents that contain terms on both sides of the operator, the intersection of both searches.
OR: Finds documents that contain either term, the union of both searches.
NOT: Finds documents that contain the term on the left but not the term on the right of the operator, the subtraction of the right hand search from the one on the left.
Please note that these Boolean Operators have to be written in uppercase to work and are processed from left to right
Phrases: Individual search terms separated by a space are joined as if an AND was put between them, unless the words match a phrase indexed by the database, in which case the phrase is searched for as written. If you want to force a search for a phrase, put the words in quotation marks “like this”. Furthermore, you can use * as a wildcard to represent any character.
Indexed Fields: Each database has various indices to improve and speed up searching - the metadata for each entry. A field can be searched specifically by putting its name in square brackets immediately after a search term. For instance, entries in Nucleotide are associated with an Organism and a Publication Date (amongst many other fields) that you can search for like so:
“Escherichia coli”[Organism] AND 2020/1/1[Publication Date]
If you want to know more about Entrez click here.
Exercise 3.4
Using NCBI search tools, find the genome record for Escherichia coli K12 MG1655.
# Genome record E.coli K12
# We start at the NCBI homepage (https://www.ncbi.nlm.nih.gov)
# Change the database to Genome
# Search for Escherichia coli K12 MG1655 (Escherichia coli K12 works too)
# An overview about Escherichia coli appears
# Scroll down to representatives
# Click on the number under RefSeq
# The genome record appears. The K12 genome has the accession number NC_000913.3
Using NCBI’s genome database, find the RefSeq reference prokaryotic genomes that are considered to have ‘Complete’ assembled genomes (there should be 15)
# Complete prokaryotic genomes
# We start at the NCBI genome page (https://www.ncbi.nlm.nih.gov/genome/)
# Select Browse by Organism
# Select prokaryotes
# Use the Filter and select under the RefSeq category reference. The 15 genomes should be selected
# The 15 genomes are:
Acinetobacter pittii PHEA-2 GCA_000191145.1
Bacillus subtilis subsp. subtilis str. 168 GCA_000009045.1
Campylobacter jejuni subsp. jejuni NCTC 11168 = ATCC 700819 GCA_000009085.1
Caulobacter vibrioides NA1000 GCA_000022005.1
Chlamydia trachomatis D/UW-3/CX GCA_000008725.1
Coxiella burnetii RSA 493 GCA_000007765.2
Escherichia coli O157:H7 str. Sakai GCA_000008865.2
Escherichia coli str. K-12 substr. MG1655 GCA_000005845.2
Klebsiella pneumoniae subsp. pneumoniae HS11286 GCA_000240185.2
Listeria monocytogenes EGD-e GCA_000196035.1
Mycobacterium tuberculosis H37Rv GCA_000195955.2
Pseudomonas aeruginosa PAO1 GCA_000006765.1
Salmonella enterica subsp. enterica serovar Typhimurium str. LT2 GCA_000006945.2
Shigella flexneri 2a str. 301 GCA_000006925.2
Staphylococcus aureus subsp. aureus NCTC 8325 GCA_000013425.1
Using NCBI’s taxonomy database, explore the record with the taxonomy 28901. At which phylogenetic level is this taxonomy given? Are there taxonomies at different phylogenetic levels?
# Explore taxonomy id 28901
# We start at the NCBI taxonomy page (https://www.ncbi.nlm.nih.gov/taxonomy/)
# Search for the taxonomy 28901
# A species-level record of Salmonella enterica appears
# If you click on this entry, a very long list of subspecies appears, thus this taxonomy is given at species level
# Click on an entry within the subspecies list (e.g. the first Salmonella enterica subsp.arizonae serovar)
# Check the taxonomy of this entry, it is *2577202*, so different than the species taxonomy
# Thus taxonomy is not specific to one genome, but is available at different phylogenetic levels
If you are in turn interested in one specific genome for your work, which type of information (instead of the taxonomy or an organism name) can you use as unique identifier?
# Find unique identifier of genome
# Continue from the subspecies entry you opened for answering the last question
# In the Entrez records at the top right corner you see how many feature entries there are
# Click on 4 next to Assembly to look at individual genomes of that subspecies
# In the results, you see 4 items (assemblies) and you see each assembly contains a Genbank accession number
# For a description of the accession number, go back to the section *Genbank flat file format*
# This Genbank accession number is unique for each genome and can be used to trace back specific genomes
# You can also use this Genbank accession number (e.g. GCA_011634505.1 from first assembly) on the NCBI homepage to search directly for a specific genome
# Note: There are a lot of different ways to find the solution. These are just examples.
Bio: a useful package¶
For this part you have to switch back once again to the terminal (Unix) as described here.
We have installed a useful package for the terminal called bio that makes the process of getting hold of sequence data much easier. You can load it as follows:
ml Bio
bio
Exercise 3.5
Find and read the available help information for bio.
# Explore the package
bio # typing bio without any arguments gives you an overview of the commands in this package
# Explore a function within the package
bio search --help # typing a command (bio search) with --help leads you to its help page (including possible arguments for the command)
# Other options to find documentation and help resources
# Check the official documentation page of the package or command
# Ask Google (type bio package documentation or any problem-related question)
# Ask Stackoverflow (https://stackoverflow.com/)
# Ask ChatGPT or use Github Copilot - but be warned that they can make easy mistakes
Choose one of the 15 genomes found in exercise 3.4 and download the fasta and genbank files using bio to your homework folder.
# For example
bio fetch GCA_000191145.1 > GCA_000191145.1.gbk
bio fasta GCA_000191145.1.gbk > GCA_000191145.1.fasta
In-class problems¶
In-class Problems Week 3
How large (in bases, or base pairs, bp) is the genome?
What is the GC content of the genome?
If the sequence has been annotated with features you may also want to know:
How many genes does the genome contain?
What are the lengths of those genes?
In many cases, you may only be interested in working with gene or protein sequences, or even a specific gene. Consider how you might write a python script that:
Outputs the DNA sequences of the genes in a genbank file to a multifasta file?
Outputs the amino acid sequences instead?
Takes as an argument a genbank file (so you can then apply it to any such file you might encounter)?
Takes as an additional argument the ID of a gene feature and outputs only that sequence?
Note: There will be no example solutions for in-class-problems. It is expected that students take notes during the lecture. If questions come up, students can use the Slack-channels to receive help.